The catabolism of amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids results in the release of nitrogen in the form of ammonium. This excess nitrogen is transported to the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy and kidneys Kidneys The kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine. Kidneys: Anatomy and eliminated from the body in the form of urea via the urine. The urea cycle (or ornithine cycle) takes place mainly in the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy and comprises the synthesis Synthesis Polymerase Chain Reaction (PCR) of urea from ammonium, CO2, aspartate Aspartate One of the non-essential amino acids commonly occurring in the l-form. It is found in animals and plants, especially in sugar cane and sugar beets. It may be a neurotransmitter. Synthesis of Nonessential Amino Acids, and bicarbonate Bicarbonate Inorganic salts that contain the -HCO3 radical. They are an important factor in determining the ph of the blood and the concentration of bicarbonate ions is regulated by the kidney. Levels in the blood are an index of the alkali reserve or buffering capacity. Electrolytes. The cycle involves 1 feeder reaction to incorporate the ammonium and 4 reactions in the cycle. It prevents cytotoxic Cytotoxic Parvovirus B19 hyperammonemia Hyperammonemia Elevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea. Cirrhosis levels.
Last updated: Apr 25, 2025
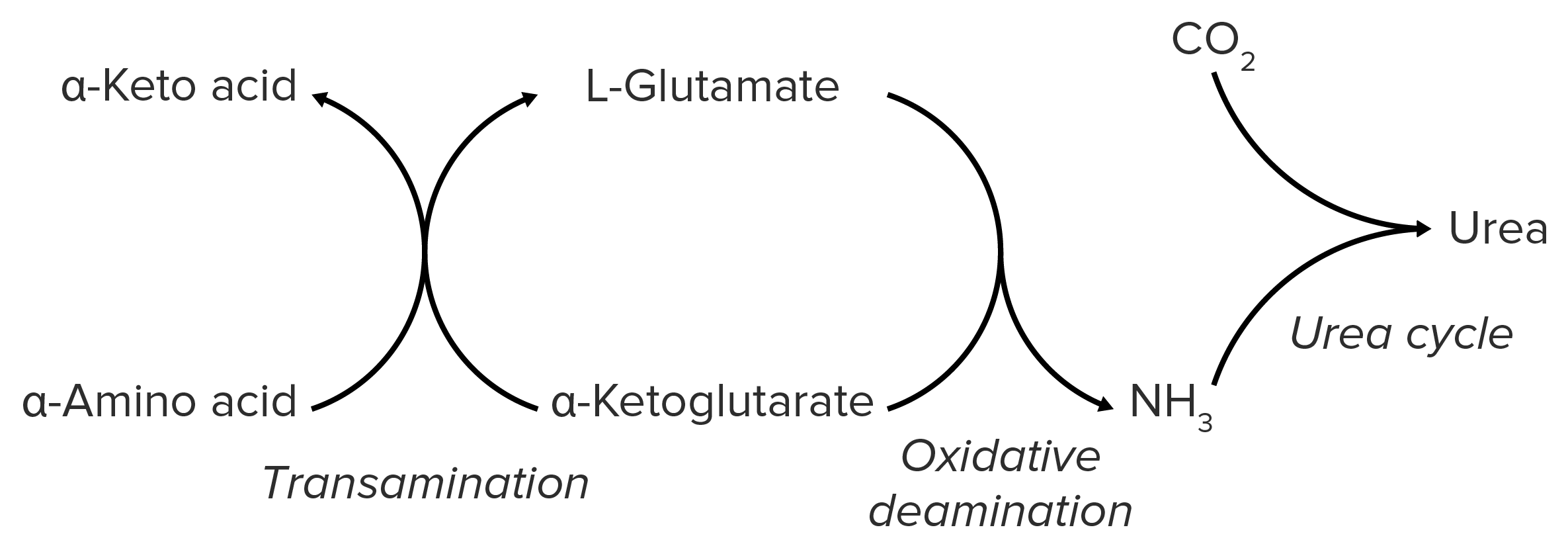
Schematic diagram of the catabolism of amino acids, resulting in the release of amino groups that are excreted from the body as urea.
Image by Lecturio.Nitrogen is produced in skeletal musculature via the catabolism of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis. It is bound to AAs in the form of amino groups → bloodstream → liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy.
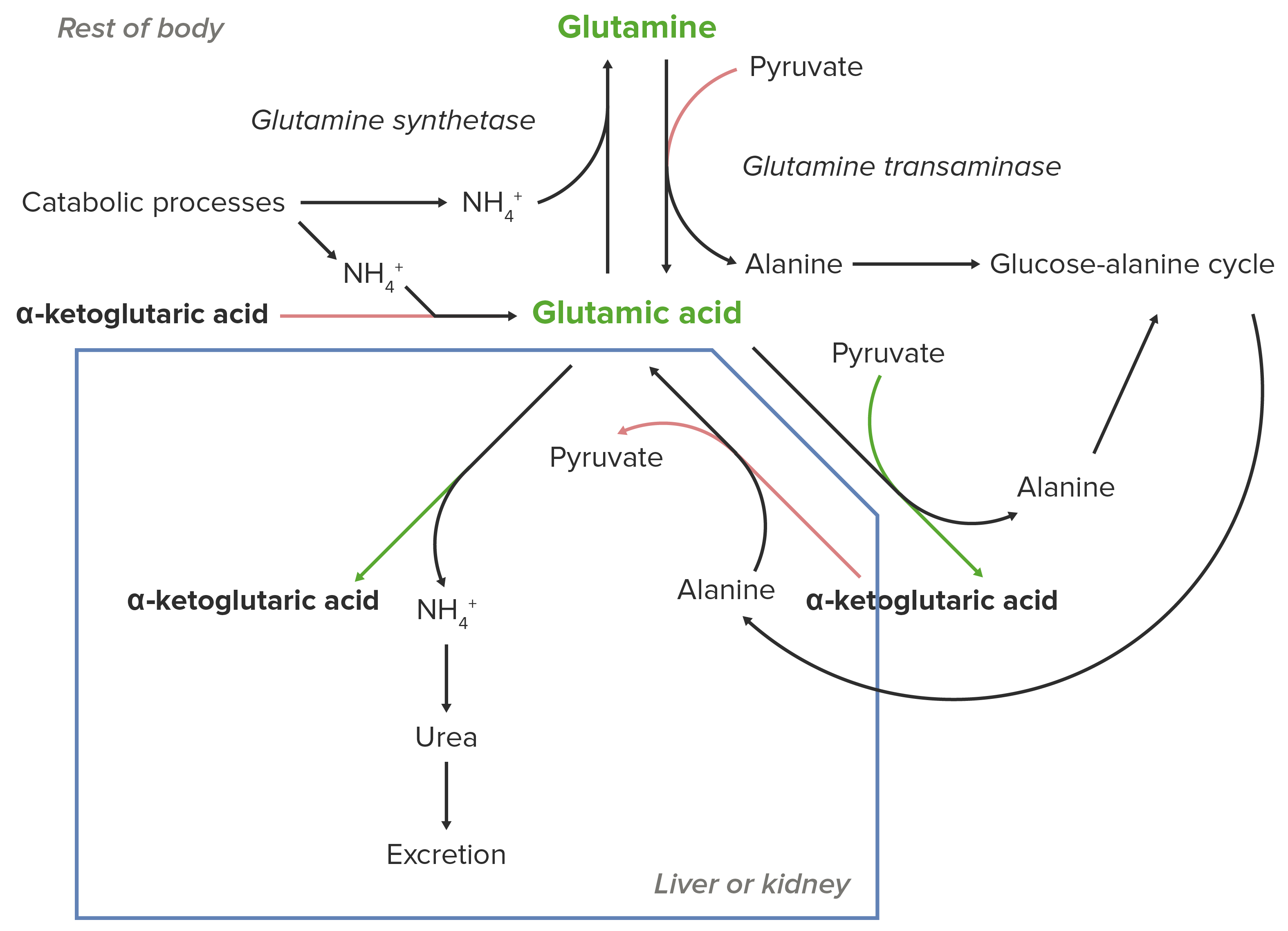
Schematic diagram of the essential role of glutamate (glutamic acid) in the transport of nitrogen from the peripheral sites of breakdown of amino acids to the liver and kidney for excretion from the body.
Image by Lecturio.Glutamate Glutamate Derivatives of glutamic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that contain the 2-aminopentanedioic acid structure. Synthesis of Nonessential Amino Acids (or glutamic acid) is essential for nitrogen transport to the kidneys Kidneys The kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine. Kidneys: Anatomy and liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy.
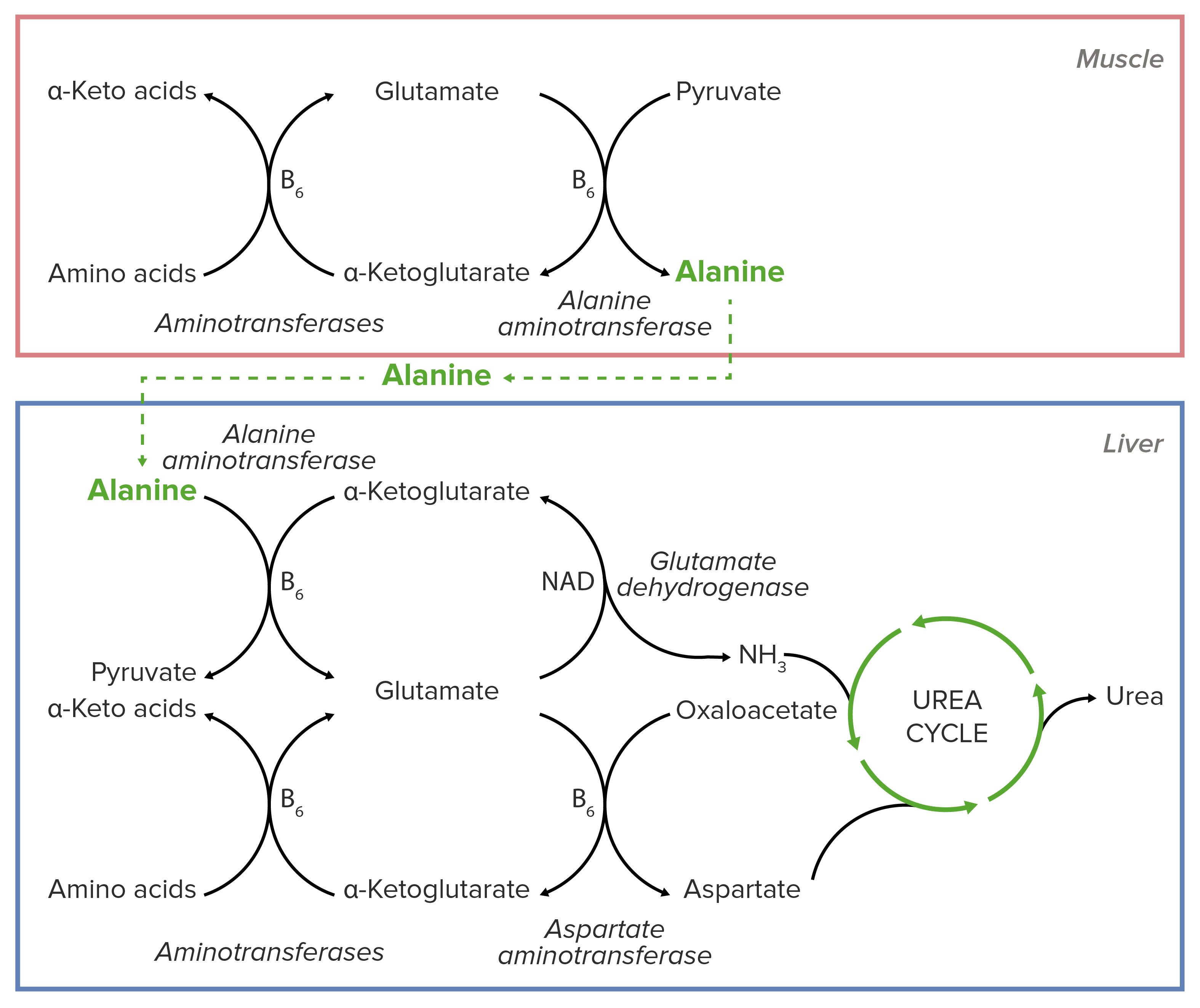
Schematic diagram of the essential role of alanine in the transport of nitrogen from muscular tissue to the liver before its introduction into the urea cycle.
Image by Lecturio.Alanine Alanine A non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system. Synthesis of Nonessential Amino Acids is essential for nitrogen transport from the muscles to the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy.
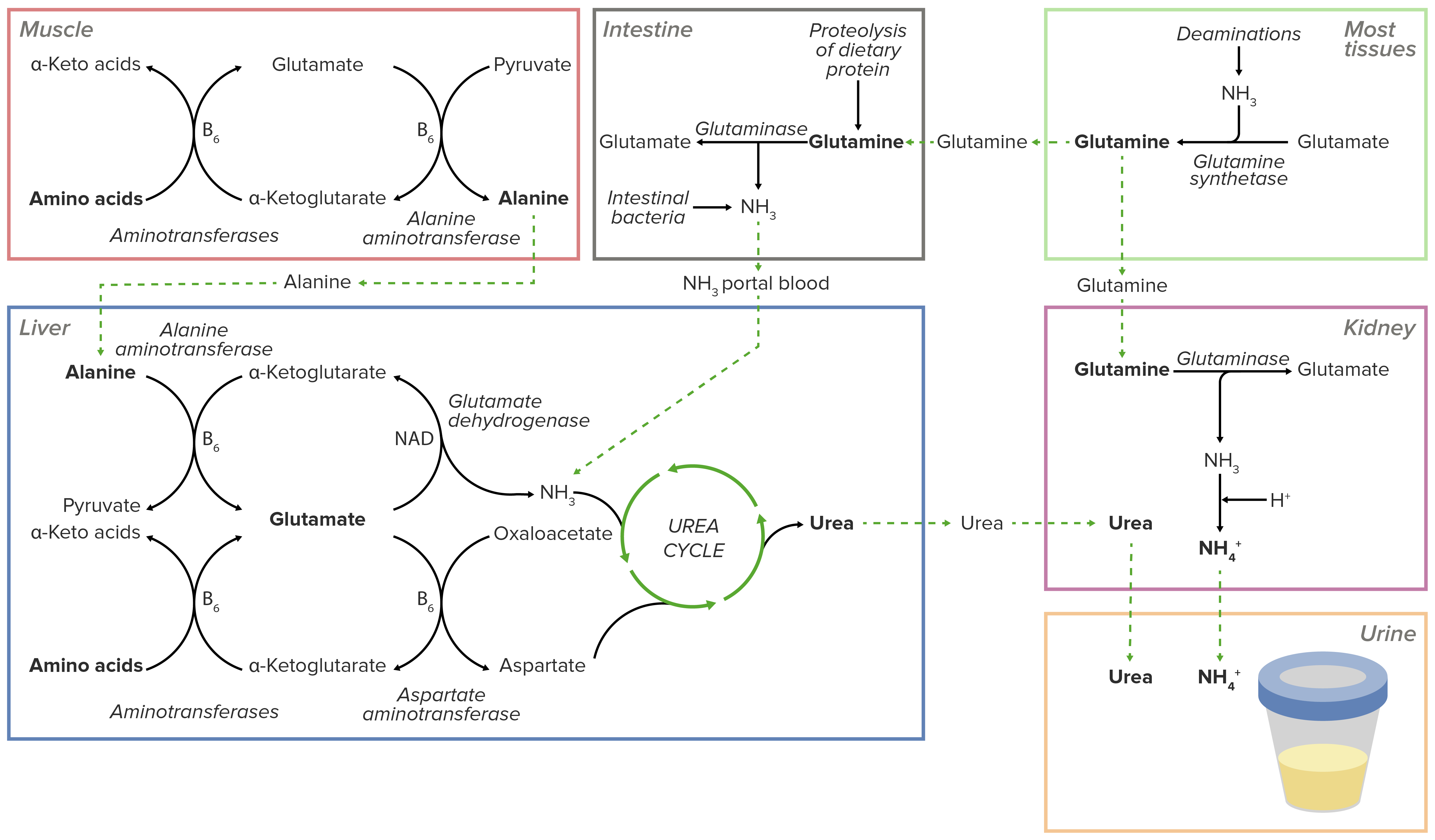
Overview of the transport of excess nitrogen in the form of amino groups to the liver and kidneys. Excess nitrogen is secreted directly into the urine in the kidneys or shunted into the urea cycle in the liver. The resulting urea is then transported to the kidneys to be excreted in the urine.
Image by Lecturio.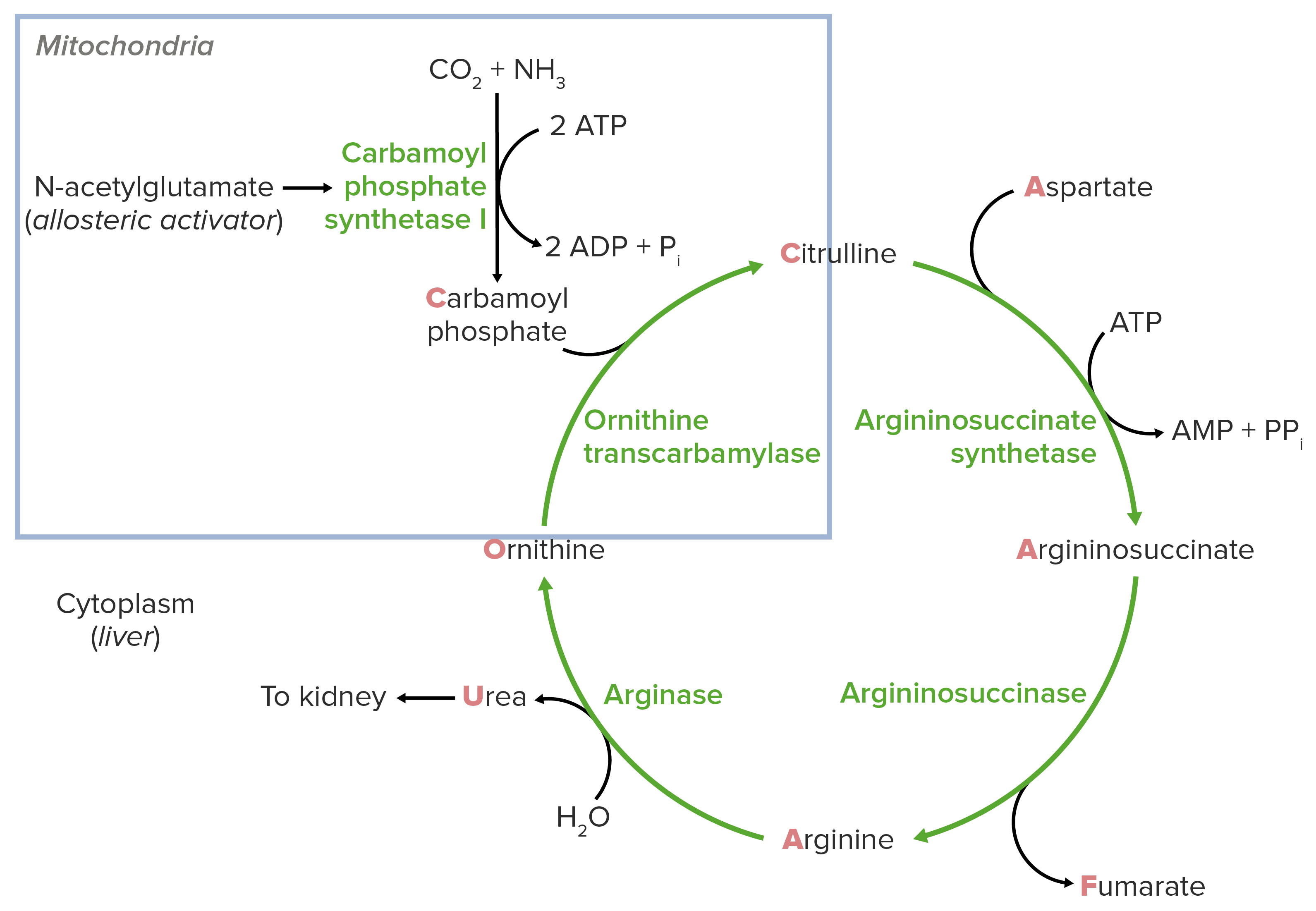
Schematic diagram of the urea cycle (feeder reaction outlined in the blue square).
Image by Lecturio.Reaction steps of the urea cycle
Congenital deficiencies of the urea cycle
Acquired conditions that affect the urea cycle