Treacher Collins syndrome is a rare genetic condition with autosomal dominant inheritance Autosomal dominant inheritance Autosomal Recessive and Autosomal Dominant Inheritance. Treacher Collins syndrome is also referred to as mandibulofacial dysostosis or Franceschetti syndrome and is characterized by significant craniofacial deformities and conductive hearing loss Conductive hearing loss Hearing loss due to interference with the mechanical reception or amplification of sound to the cochlea. The interference is in the outer or middle ear involving the ear canal; tympanic membrane; or ear ossicles. Hearing Loss. Treacher Collins syndrome is strictly a physical disease and does not affect cognition or other spheres of development. Diagnosis is confirmed with genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Management of the airway Airway ABCDE Assessment, feeding ability, and hearing loss Hearing loss Hearing loss, also known as hearing impairment, is any degree of impairment in the ability to apprehend sound as determined by audiometry to be below normal hearing thresholds. Clinical presentation may occur at birth or as a gradual loss of hearing with age, including a short-term or sudden loss at any point. Hearing Loss are the primary concerns in the first few years of life. Ultimately, facial reconstructive surgery is needed. Life expectancy Life expectancy Based on known statistical data, the number of years which any person of a given age may reasonably expected to live. Population Pyramids is normal.
Last updated: Dec 15, 2025
This syndrome is characterized by a multitude of bilateral and often asymmetric craniofacial structural defects and abnormalities.
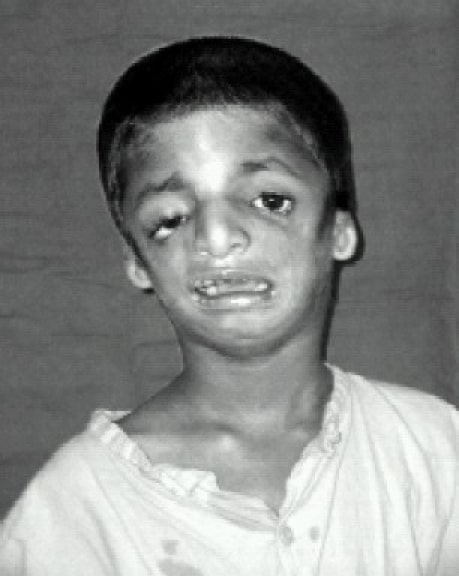
Patient showing hypoplasia of facial bones, down-sloping palpebral fissures, and coloboma of the lower eyelids with scanty lower eyelashes
Image: “Front view” by Indian J Anaesth. License: CC BY 2.0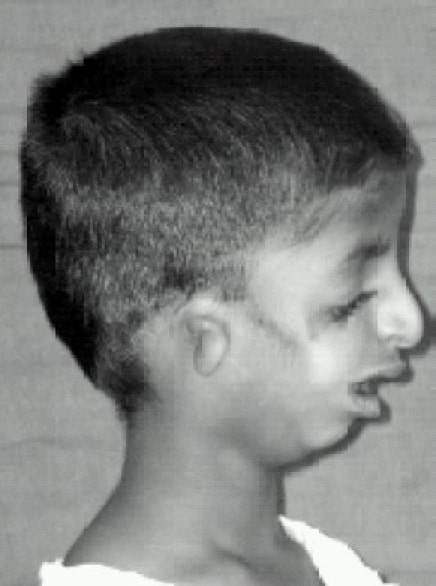
Lateral view of the patient showing deformity of the ear (microtia) and retrognathia
Image: “Lateral view” by Indian J Anaesth. License: CC BY 2.0Craniofacial malformations can result in airway Airway ABCDE Assessment compromise and feeding difficulties, making management of these problems the priority in early years. Once life-threatening issues are resolved, management of hearing loss Hearing loss Hearing loss, also known as hearing impairment, is any degree of impairment in the ability to apprehend sound as determined by audiometry to be below normal hearing thresholds. Clinical presentation may occur at birth or as a gradual loss of hearing with age, including a short-term or sudden loss at any point. Hearing Loss is imperative for speech development. Surgery is needed for correction of facial abnormalities.