Hirschsprung disease (HD), also known as congenital aganglionosis or congenital megacolon Megacolon Megacolon is a severe, abnormal dilatation of the colon, and is classified as acute or chronic. There are many etiologies of megacolon, including neuropathic and dysmotility conditions, severe infections, ischemia, and inflammatory bowel disease. Megacolon, is a congenital anomaly of the colon Colon The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy caused by the failure of neural crest-derived ganglion cells Ganglion Cells The Visual Pathway and Related Disorders to migrate into the distal colon Colon The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy. The lack of innervation always involves the rectum Rectum The rectum and anal canal are the most terminal parts of the lower GI tract/large intestine that form a functional unit and control defecation. Fecal continence is maintained by several important anatomic structures including rectal folds, anal valves, the sling-like puborectalis muscle, and internal and external anal sphincters. Rectum and Anal Canal: Anatomy and extends proximally and contiguously over variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables distances. Most cases are diagnosed in the neonatal period, with a classic triad of symptoms including delayed passage of meconium Meconium The thick green-to-black mucilaginous material found in the intestines of a full-term fetus. It consists of secretions of the intestinal glands; bile pigments; fatty acids; amniotic fluid; and intrauterine debris. It constitutes the first stools passed by a newborn. Prenatal and Postnatal Physiology of the Neonate, abdominal distension, and bilious vomiting Bilious Vomiting Congenital Duodenal Obstruction. Individuals having less severe degrees of functional obstruction may not be diagnosed until later in infancy or childhood when they present with symptoms of chronic refractory constipation Constipation Constipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation, abdominal distension, and failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive. The diagnosis of HD is confirmed by the absence of ganglion cells Ganglion Cells The Visual Pathway and Related Disorders on rectal biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma after noninvasive testing such as anorectal manometry Anorectal Manometry Pediatric Constipation and the use of contrast enema. Surgical resection of the aganglionic segment is the standard treatment.
Last updated: Dec 15, 2025
Hirschsprung disease (HD), also known as congenital aganglionosis or congenital megacolon Megacolon Megacolon is a severe, abnormal dilatation of the colon, and is classified as acute or chronic. There are many etiologies of megacolon, including neuropathic and dysmotility conditions, severe infections, ischemia, and inflammatory bowel disease. Megacolon, is a congenital anomaly of the colon Colon The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy caused by the failure of neural crest-derived ganglion cells Ganglion Cells The Visual Pathway and Related Disorders to migrate into the distal colon Colon The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy.
The pathophysiology in HD is the complete absence of ganglion cells Ganglion Cells The Visual Pathway and Related Disorders (aganglionosis) in the intrinsic nerve supply of the bowel due to the failure of neural crest-derived ganglion cells Ganglion Cells The Visual Pathway and Related Disorders to migrate completely into the myenteric plexuses of the wall of the colon Colon The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy.
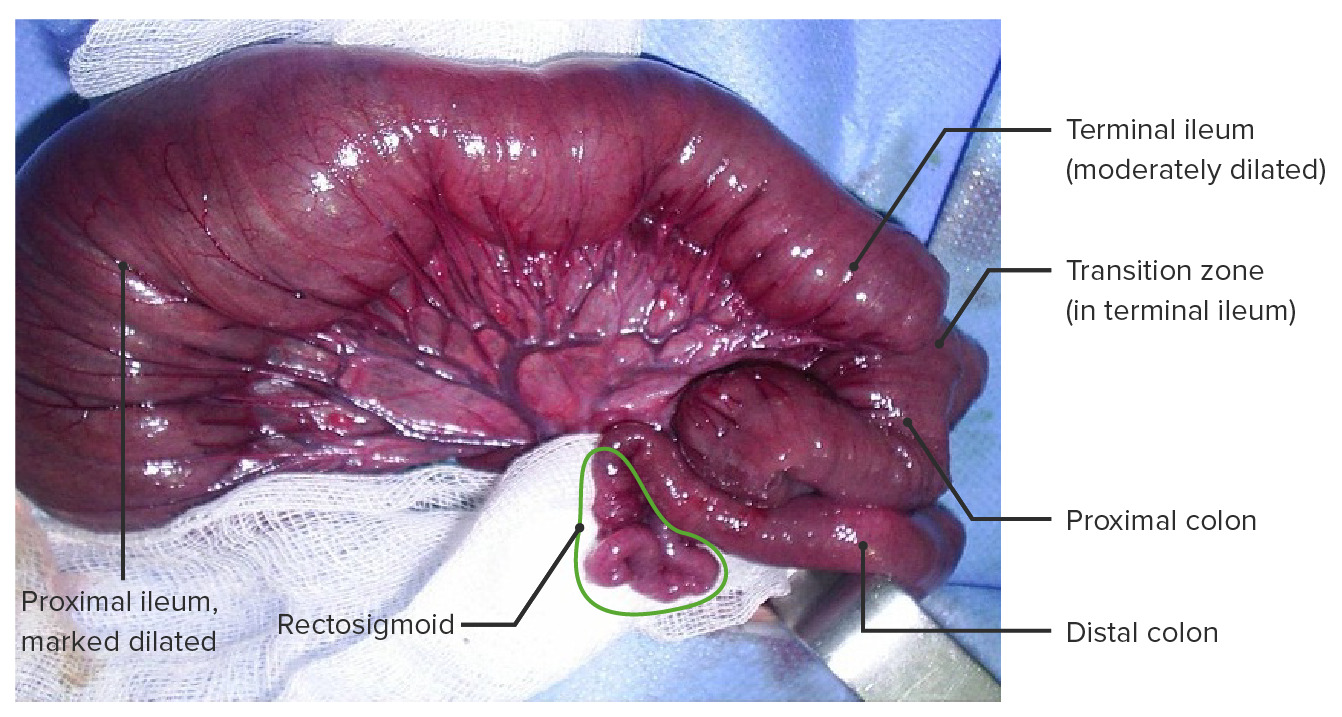
Hirschsprung disease with total colonic aganglionosis including a short segment of the terminal ileum.
This type of aganglionosis occurs in < 5% of cases.
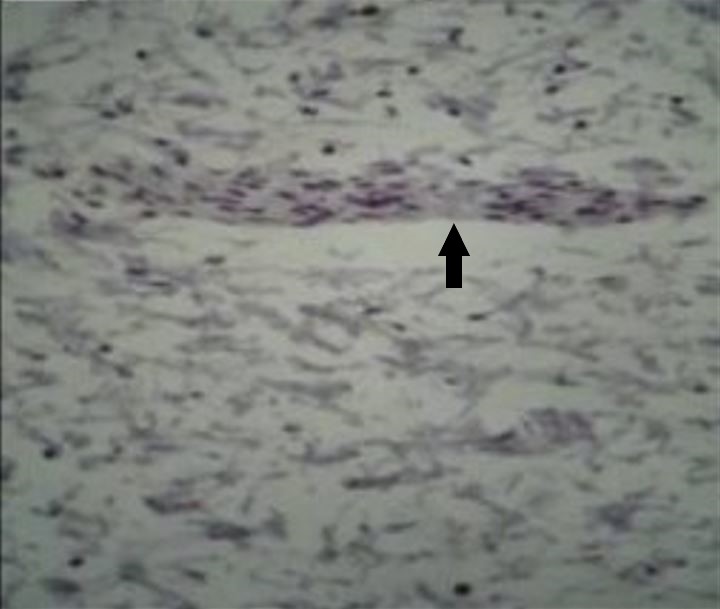
Microphotograph of Hirschsprung disease (HD)-affected segment of intestine stained with the immunohistochemical (IHC) marker calretinin, which shows no brown staining of ganglion cells within a nerve (arrow) in the myenteric plexus
Image: “Total absence of staining after calretinin immunohistochemistry in the aganglionic segment” by Hiradfar M, Sharifi N, Khajedaluee M, Zabolinejad N, Taraz Jamshidi S. License: CC BY 3.0, edited by Lecturio.Most individuals with HD are diagnosed in the 1st month of life. Individuals with less severe disease may not present with symptoms until 3 years of age in approximately 10% of cases.
If HD is suspected based on the clinical presentations in the neonatal or postnatal periods, the following diagnostic tests Diagnostic tests Diagnostic tests are important aspects in making a diagnosis. Some of the most important epidemiological values of diagnostic tests include sensitivity and specificity, false positives and false negatives, positive and negative predictive values, likelihood ratios, and pre-test and post-test probabilities. Epidemiological Values of Diagnostic Tests are used:
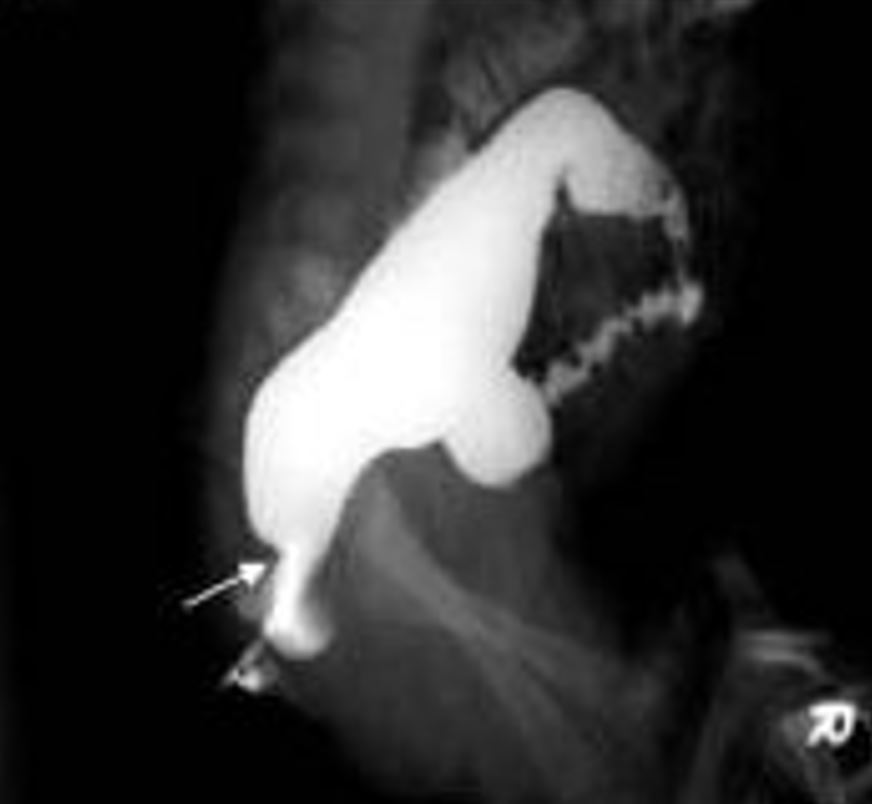
Hirschsprung disease as seen on contrast barium enema study, with the arrow showing the “transition zone” between the normal and aganglionic bowel
Image: “Contrast enema showing a CETZ at rectosigmoid, arrow” by Pratap, A., et al. License: CC BY 2.0, cropped by Lecturio.Causes of intestinal obstruction Intestinal obstruction Any impairment, arrest, or reversal of the normal flow of intestinal contents toward the anal canal. Ascaris/Ascariasis in the neonatal period can be distinguished from HD based on their clinical features and the presence of ganglia on a rectal biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma.
Causes of intestinal obstruction Intestinal obstruction Any impairment, arrest, or reversal of the normal flow of intestinal contents toward the anal canal. Ascaris/Ascariasis in older infants and children: