Selective immunoglobulin A ( IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions) deficiency is the most common type of primary immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome. The condition is a hypogammaglobulinemia characterized by a lack or reduced levels of IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions. This antibody mainly resides in the mucous membranes of the mouth, airways, and digestive tract. The exact cause is unknown. The disease is usually asymptomatic, although some patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship can present with recurrent respiratory and gastrointestinal infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease as well as autoimmune and malignant disorders. Diagnosis is made with a measure of exceptionally low IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions levels in the serum in the presence of normal IgG IgG The major immunoglobulin isotype class in normal human serum. There are several isotype subclasses of igg, for example, igg1, igg2a, and igg2b. Hypersensitivity Pneumonitis and IgM IgM A class of immunoglobulin bearing mu chains (immunoglobulin mu-chains). Igm can fix complement. The name comes from its high molecular weight and originally being called a macroglobulin. Immunoglobulins: Types and Functions levels.
Last updated: Dec 15, 2025
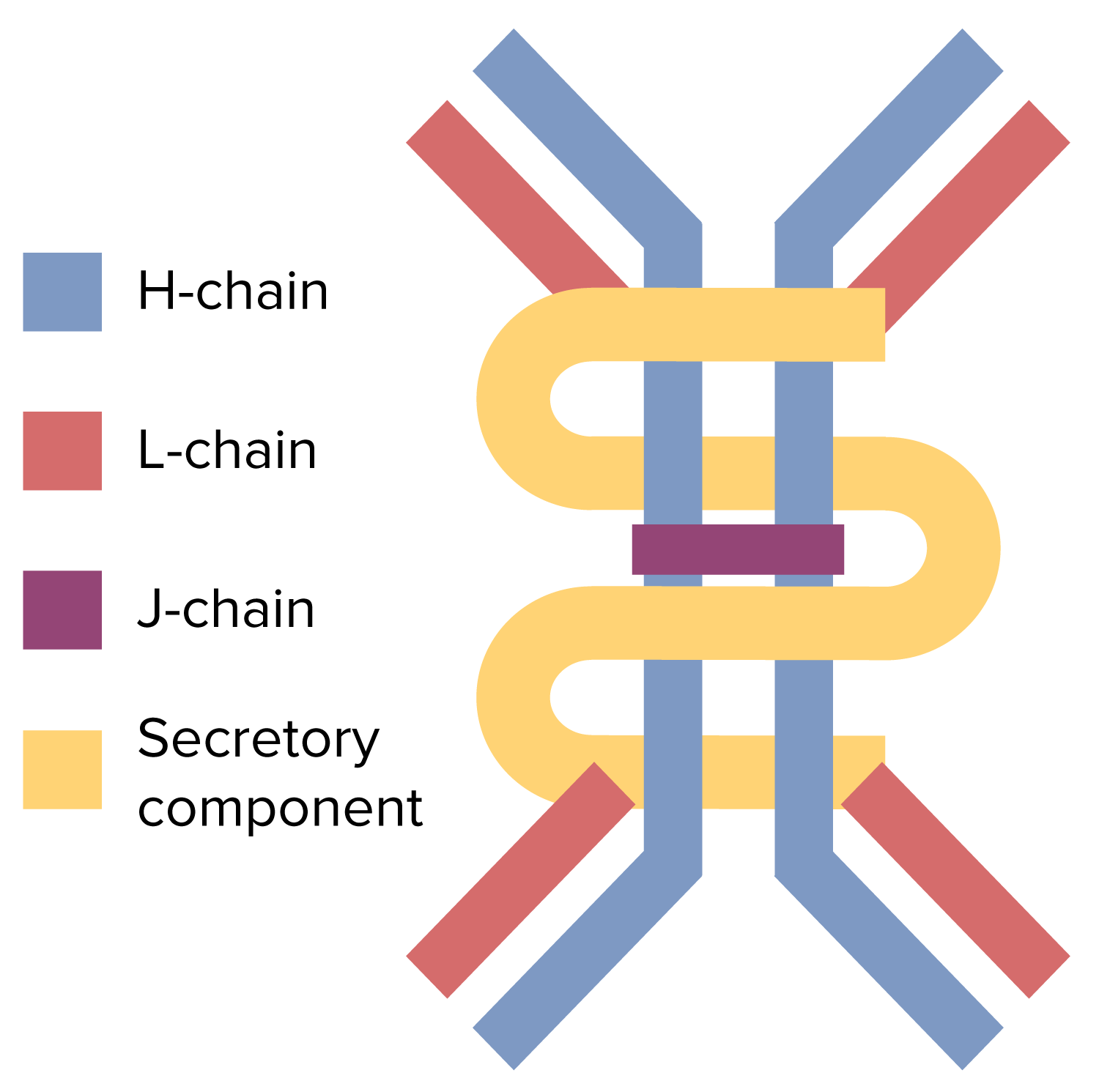
The dimeric structure of the IgA antibody
Image by Lecturio.Approximately 90% of cases are asymptomatic.
Symptomatic patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship may present a mixture of the following symptoms:
Diagnosis
Management
The following conditions are other congenital B-cell immunodeficiencies that serve as differential diagnoses for selective IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions deficiency.