Severe combined immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome (SCID), also called “bubble boy disease,” is a rare genetic disorder in which the development of functional B and T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified - cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions is disturbed due to several genetic mutations Genetic Mutations Carcinogenesis that result in reduced or absent immune function. It is the most severe form of primary immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome and is characterized by dysfunction in both humoral and cell-mediated immune responses. Multiple mutations can result in heterogeneous types of SCID. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with severe and recurrent infections Recurrent infections Common Variable Immunodeficiency (CVID) within the first months of life. Management includes IV immunoglobulins IV immunoglobulins Immunoglobulin preparations used in intravenous infusion, containing primarily immunoglobulin g. They are used to treat a variety of diseases associated with decreased or abnormal immunoglobulin levels including pediatric aids; primary hypergammaglobulinemia; scid; cytomegalovirus infections in transplant recipients, lymphocytic leukemia, chronic; kawasaki syndrome, infection in neonates, and idiopathic thrombocytopenic purpura. DiGeorge Syndrome and bone marrow transplantation Bone marrow transplantation Transfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms. Organ Transplantation. If left untreated, SCID is usually fatal within the 1st year of life.
Last updated: Dec 15, 2025
At least 12 genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure are known to cause severe combined immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome (SCID) if mutated, including those that encode for the following:
The pattern of inheritance depends on the type of genetic mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations involved:
| Phenotype Phenotype The complete genetic complement contained in the DNA of a set of chromosomes in a human. The length of the human genome is about 3 billion base pairs. Basic Terms of Genetics | Genetic defect Genetic Defect Ion Channel Myopathy |
|---|---|
| T–B+NK– | γC, JAK3 |
| T–B–NK+ | RAG-1, RAG-2, Artemis, DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure ligase IV, Cernunnos, DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure PKcs |
| T–B+NK+ | IL-7Rα, CD3δ, CD3ζ, Coronin-1A, ZAP-70, CD45 |
| T–B–NK– | ADA ADA An enzyme that catalyzes the hydrolysis of adenosine to inosine with the elimination of ammonia. Purine and Pyrimidine Metabolism, AK2 |
| Syndrome | Defect |
|---|---|
| X-linked X-linked Genetic diseases that are linked to gene mutations on the X chromosome in humans or the X chromosome in other species. Included here are animal models of human X-linked diseases. Common Variable Immunodeficiency (CVID) severe combined immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome | Mutations in the gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics encoding the common γ chain, a protein that is shared by the receptors Receptors Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors for interleukins Interleukins Interleukins are a type of cytokines (signaling proteins) that communicate messages between different parts of the immune system. The majority of interleukins are synthesized by helper CD4 T lymphocytes along with other cells such as monocytes, macrophages, and endothelial cells. Interleukins IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 |
| Adenosine Adenosine A nucleoside that is composed of adenine and d-ribose. Adenosine or adenosine derivatives play many important biological roles in addition to being components of DNA and RNA. Adenosine itself is a neurotransmitter. Class 5 Antiarrhythmic Drugs deaminase deficiency |
|
| Purine nucleoside phosphorylase Purine nucleoside phosphorylase An enzyme that catalyzes the reaction between a purine nucleoside and orthophosphate to form a free purine plus ribose-5-phosphate. Purine and Pyrimidine Metabolism ( PNP PNP An enzyme that catalyzes the reaction between a purine nucleoside and orthophosphate to form a free purine plus ribose-5-phosphate. Purine and Pyrimidine Metabolism) deficiency | Autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance disorder involving mutations of the PNP PNP An enzyme that catalyzes the reaction between a purine nucleoside and orthophosphate to form a free purine plus ribose-5-phosphate. Purine and Pyrimidine Metabolism gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics |
| Reticular dysgenesis | Inability of granulocyte precursors to form granules secondary to mitochondrial adenylate kinase 2 malfunction |
| Omenn syndrome Omenn syndrome An autosomal recessive form of severe combined immunodeficiency (SCID) caused by mutations in the RAG1 or RAG2 genes. IPEX Syndrome |
|
| Bare lymphocyte syndrome |
|
| JAK3 | JAK3 is an enzyme that mediates transduction Transduction The transfer of bacterial DNA by phages from an infected bacterium to another bacterium. This also refers to the transfer of genes into eukaryotic cells by viruses. This naturally occurring process is routinely employed as a gene transfer technique. Bacteriology downstream of the γc signals. |
The disease typically presents in early childhood (2–6 months). Severe and recurrent opportunistic infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease are seen in affected individuals.
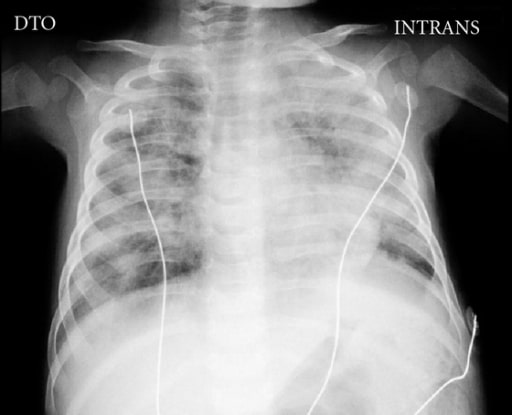
Chest X-ray of a 5-month-old boy with SCID complicated by disseminated bacillus Calmette-Guérin (BCG) disease showing the absence of a thymus and bilateral areas of opacities
Image: “Chest X-ray” by Pediatric Intensive Care Unit, Hospital Dona Estefânia, 1169-045 Lisboa, Portugal. License: CC BY 3.0
Extreme measures were historically used to achieve efficient isolation of SCID patients, earning SCID the name “bubble boy disease.”
Image: “David Vetter and John Montgomery” by Jeremy112233. License: Public DomainThe following conditions are differential diagnoses for SCID: