Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) is a subcategory of the vasculitides that includes granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA), renal-limited necrotizing and crescentic glomerulonephritis, and drug-induced ANCA-associated vasculitis. All 5 diseases may cause life-threatening small-vessel vasculitis with a wide range of systemic manifestations, which can involve the lungs, kidneys, skin, heart, and other tissues. Diagnosis is suspected by clinical presentation and a positive cytoplasmic (c)- or perinuclear (p)-ANCA test. Biopsy of involved tissue confirms the diagnosis. Glucocorticoids and immunosuppressive therapy are the mainstays of treatment.
Antineutrophil cytoplasmic antibody (ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis)-associated vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus (AAV) is characterized by necrotizing small-vessel vasculitisSmall-Vessel VasculitisHenoch-Schönlein Purpura and a positive ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis test without significant immune complex deposition.
Background
Antineutrophil cytoplasmic antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions (ANCAs) are:
AutoantibodiesAutoantibodiesAntibodies that react with self-antigens (autoantigens) of the organism that produced them.Blotting Techniques directed against proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis contained within neutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation (and monocytesMonocytesLarge, phagocytic mononuclear leukocytes produced in the vertebrate bone marrow and released into the blood; contain a large, oval or somewhat indented nucleus surrounded by voluminous cytoplasm and numerous organelles.Innate Immunity: Phagocytes and Antigen Presentation, to a lesser degree)
Specific to protein targets associated with cytoplasmic granules of neutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation and, on binding, triggerTriggerThe type of signal that initiates the inspiratory phase by the ventilatorInvasive Mechanical Ventilation an exaggerated inflammatory response (i.e., an autoimmune reaction)
There are 2 clinically significant subtypes of ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis:
BindBINDHyperbilirubinemia of the Newborn to MPOMPOAcute Myeloid Leukemia (there are less clinically significant pANCA autoantibodiesAutoantibodiesAntibodies that react with self-antigens (autoantigens) of the organism that produced them.Blotting Techniques that target elastaseElastaseA protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin.Proteins and Peptides, cathepsin G, lysozyme, and lactoferrin).
MPOMPOAcute Myeloid Leukemia is a protein found in neutrophil granules that is involved in the formation of reactive oxygen speciesReactive oxygen speciesMolecules or ions formed by the incomplete one-electron reduction of oxygen. These reactive oxygen intermediates include singlet oxygen; superoxides; peroxides; hydroxyl radical; and hypochlorous acid. They contribute to the microbicidal activity of phagocytes, regulation of signal transduction and gene expression, and the oxidative damage to nucleic acids; proteins; and lipids.Metabolic Dysfunction-associated Steatotic Liver Disease (MASLD) for antimicrobial purposes.
MPOMPOAcute Myeloid Leukemia deficiency (secondary to autoimmune neutrophil destruction) results in an exaggerated inflammatory response.
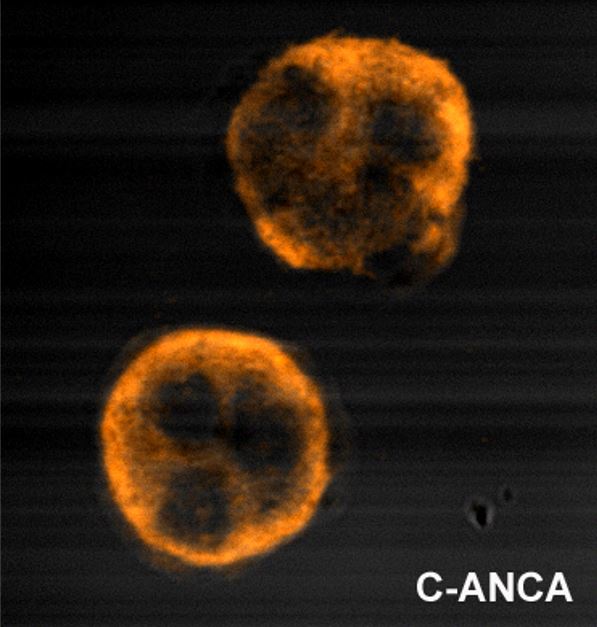
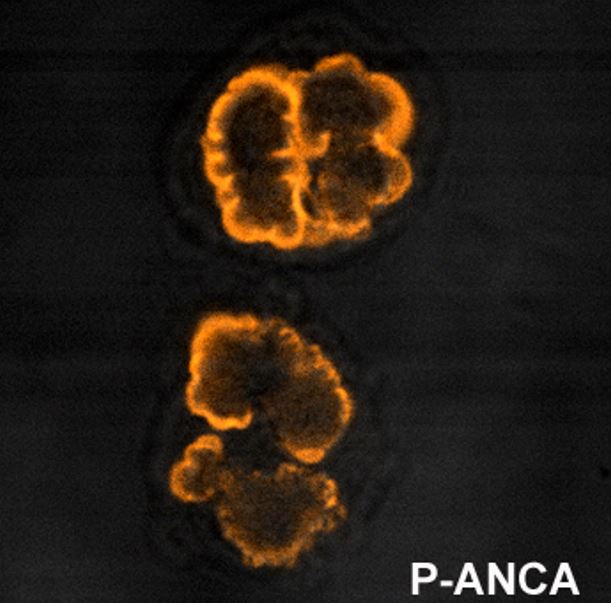
The “p” in pANCA stands for perinuclear because on staining, these antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions stain the perinuclear area of the neutrophil (where MPOMPOAcute Myeloid Leukemia and other negatively charged proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are highly concentrated).
PR3 deficiency (secondary to autoimmune neutrophil destruction) results in an exaggerated inflammatory response.
The “c” in cANCA stands for cytoplasmic because on staining, these antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions stain the peripheral cytoplasmic area of the neutrophil (where PR3, which is positively charged, is highly concentrated).
The presence of ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis (i.e., a positive ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis test):
Does not:
Diagnose a disease entity
Correlate with symptom severity
Correlate with disease activity
Is simply a marker that helps establish patterns of disease
ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis positivity is associated with autoimmune disease processes (the following pANCA/cANCA designations are only a guide; significant variability exists):
VasculitidesVasculitidesVasculitides are a group of conditions characterized by vasculitis, ischemia, and damage to the organs supplied by the affected vessels. The affected arteries are of different sizes and locations and vary by the type of vasculitis. Vasculitides:
Granulomatosis with polyangiitisGranulomatosis with PolyangiitisA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis (cANCA)
Microscopic polyangiitisMicroscopic polyangiitisA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides (pANCA)
Eosinophilic granulomatosis with polyangiitisGranulomatosis with PolyangiitisA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis (pANCA)
Primary pauci-immune necrotizing crescenteric glomerulonephritis (aka rapidly progressive glomerulonephritisRapidly Progressive GlomerulonephritisRapidly progressive glomerulonephritis (RPGN) is a syndrome of severe glomerular disease with progressive loss of kidney function within weeks to months. Histologically, crescents (the proliferation of epithelial cells and the infiltration of monocytes/macrophages in the Bowman space) are found in the glomeruli and arise from immunologic injury.Rapidly Progressive Glomerulonephritis (RPGNRPGNRapidly progressive glomerulonephritis (RPGN) is a syndrome of severe glomerular disease with progressive loss of kidney function within weeks to months. Histologically, crescents (the proliferation of epithelial cells and the infiltration of monocytes/macrophages in the Bowman space) are found in the glomeruli and arise from immunologic injury.Rapidly Progressive Glomerulonephritis) or renal-limited vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus) (pANCA)
Drug-induced ANCA-associated vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus (pANCA)
Anti–glomerular basement membraneBasement membraneA darkly stained mat-like extracellular matrix (ecm) that separates cell layers, such as epithelium from endothelium or a layer of connective tissue. The ecm layer that supports an overlying epithelium or endothelium is called basal lamina. Basement membrane (bm) can be formed by the fusion of either two adjacent basal laminae or a basal lamina with an adjacent reticular lamina of connective tissue. Bm, composed mainly of type IV collagen; glycoprotein laminin; and proteoglycan, provides barriers as well as channels between interacting cell layers.Thin Basement Membrane Nephropathy (TBMN) (GBM) autoantibody disease (pANCA)
Interstitial nephritis (pANCA)
Ulcerative colitisColitisInflammation of the colon section of the large intestine, usually with symptoms such as diarrhea (often with blood and mucus), abdominal pain, and fever.Pseudomembranous Colitis (pANCA)
Primary sclerosing cholangitisPrimary Sclerosing CholangitisPrimary sclerosing cholangitis (PSC) is an inflammatory disease that causes fibrosis and strictures of the bile ducts. The exact etiology is unknown, but there is a strong association with IBD. Patients typically present with an insidious onset of fatigue, pruritus, and jaundice, which can progress to cirrhosis and complications related to biliary obstruction. Primary Sclerosing Cholangitis (pANCA)
Autoimmune hepatitisAutoimmune hepatitisAutoimmune hepatitis (AIH) is a rare form of chronic liver disease in which the immune system attacks the liver causing inflammation. It predominantly affects women. Clinical presentation ranges from asymptomatic cases to patients that present with symptoms of acute liver failure (jaundice, right upper quadrant pain).Autoimmune Hepatitis (pANCA)
Granulomatosis with polyangiitisGranulomatosis with PolyangiitisA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis (GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis):
The majority of cases are cANCA+ (80%–90%).
A small percentage are pANCA+ or ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis–.
Microscopic polyangiitisMicroscopic polyangiitisA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides (MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides):
The majority of cases are pANCA+ (nearly 90%).
A small percentage are cANCA+ or ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis–.
Eosinophilic granulomatosis with polyangiitisGranulomatosis with PolyangiitisA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis (EGPA; also known as Churg-Strauss syndromeChurg-Strauss syndromeWidespread necrotizing angiitis with granulomas. Pulmonary involvement is frequent. Asthma or other respiratory infection may precede evidence of vasculitis. Eosinophilia and lung involvement differentiate this disease from polyarteritis nodosa.Vasculitides):
The majority of cases are ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis– (50%–70%).
A minority of cases are ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis+ (30%–50%).
The majority of ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis+ cases are pANCA+ (75%–80%).
A small percentage are cANCA+.
Some sources also include the following 2 diseases:
Renal-limited necrotizing and crescentic glomerulonephritis:
The majority of cases are pANCA+ (75%–80%).
A small percentage are cANCA+ or ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis–.
Drug-induced ANCA-associated vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus:
The majority of cases are pANCA+.
A small percentage are cANCA+.
Table: Drugs implicated in ANCA-associated vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus
Antithyroid agentsAntithyroid agentsAntithyroid agents are used in the management of hyperthyroidism, particularly that due to Graves’ disease. This group of medications includes the thionamides (methimazole and propylthiouracil), potassium iodide, and radioactive iodine. Thionamides are currently the preferred choice for hyperthyroid management.Antithyroid Drugs
PropylthiouracilPropylthiouracilA thiourea antithyroid agent. Propylthiouracil inhibits the synthesis of thyroxine and inhibits the peripheral conversion of thyroxine to triiodothyronine. It is used in the treatment of hyperthyroidism.Antithyroid Drugs
MethimazoleMethimazoleA thioureylene antithyroid agent that inhibits the formation of thyroid hormones by interfering with the incorporation of iodine into tyrosyl residues of thyroglobulin. This is done by interfering with the oxidation of iodide ion and iodotyrosyl groups through inhibition of the peroxidase enzyme.Antithyroid Drugs
Biologics
EtanerceptEtanerceptA recombinant version of soluble human tnf receptor fused to an IgG Fc fragment that binds specifically to tumor necrosis factor and inhibits its binding with endogenous tnf receptors. It prevents the inflammatory effect of tnf and is used to treat rheumatoid arthritis; psoriatic arthritis and ankylosing spondylitis.Immunosuppressants
InfliximabInfliximabA chimeric monoclonal antibody to tnf-alpha that is used in the treatment of rheumatoid arthritis; ankylosing spondylitis; psoriatic arthritis and Crohn’s disease.Disease-Modifying Antirheumatic Drugs (DMARDs)
AdalimumabAdalimumabA humanized monoclonal antibody that binds specifically to tnf-alpha and blocks its interaction with endogenous tnf receptors to modulate inflammation. It is used in the treatment of rheumatoid arthritis; psoriatic arthritis; Crohn’s disease and ulcerative colitis.Disease-Modifying Antirheumatic Drugs (DMARDs)
Antimicrobials
TrimethoprimTrimethoprimThe sulfonamides are a class of antimicrobial drugs inhibiting folic acid synthesize in pathogens. The prototypical drug in the class is sulfamethoxazole. Although not technically sulfonamides, trimethoprim, dapsone, and pyrimethamine are also important antimicrobial agents inhibiting folic acid synthesis. The agents are often combined with sulfonamides, resulting in a synergistic effect. Sulfonamides and Trimethoprim–sulfamethoxazoleSulfamethoxazoleA bacteriostatic antibacterial agent that interferes with folic acid synthesis in susceptible bacteria. Its broad spectrum of activity has been limited by the development of resistance.Sulfonamides and Trimethoprim (TMP-SMX)
IsoniazidIsoniazidAntibacterial agent used primarily as a tuberculostatic. It remains the treatment of choice for tuberculosis.Antimycobacterial Drugs
RifampinRifampinA semisynthetic antibiotic produced from streptomyces mediterranei. It has a broad antibacterial spectrum, including activity against several forms of Mycobacterium. In susceptible organisms it inhibits dna-dependent RNA polymerase activity by forming a stable complex with the enzyme. It thus suppresses the initiation of RNA synthesis. Rifampin is bactericidal, and acts on both intracellular and extracellular organisms.Epiglottitis
VancomycinVancomycinAntibacterial obtained from streptomyces orientalis. It is a glycopeptide related to ristocetin that inhibits bacterial cell wall assembly and is toxic to kidneys and the inner ear.Glycopeptides
MinocyclineMinocyclineA tetracycline analog, having a 7-dimethylamino and lacking the 5 methyl and hydroxyl groups, which is effective against tetracycline-resistant staphylococcus infections.Tetracyclines
Nitrofurantoin
Rheumatologic agents
AllopurinolAllopurinolA xanthine oxidase inhibitor that decreases uric acid production. It also acts as an antimetabolite on some simpler organisms.Gout Drugs
SulfasalazineSulfasalazineA drug that is used in the management of inflammatory bowel diseases. Its activity is generally considered to lie in its metabolic breakdown product, 5-aminosalicylic acid released in the colon.Sulfonamides and Trimethoprim
D-penicillamineD-penicillamineThe most characteristic degradation product of the penicillin antibiotics. It is used as an antirheumatic and as a chelating agent in wilson’s disease.Movement Disorder Drugs
AntihypertensivesAntihypertensivesThe 1st-line medication classes for hypertension include thiazide-like diuretics, angiotensin-converting enzyme inhibitors (ACEis), angiotensin II receptor blockers (ARBs), and calcium channel blockers (CCBS). Contraindications, adverse effects, and drug-to-drug interactions are agent specific.Hypertension Drugs
AtorvastatinAtorvastatinA pyrrole and heptanoic acid derivative, hydroxymethylglutaryl-CoA reductase inhibitor (statin), and anticholesteremic agent that is used to reduce serum levels of ldl-cholesterol; apolipoprotein b; and triglycerides. It is used to increase serum levels of hdl-cholesterol in the treatment of hyperlipidemias, and for the prevention of cardiovascular diseases in patients with multiple risk factors.Statins
Neurologic/psychiatric agents
ClozapineClozapineA tricyclic dibenzodiazepine, classified as an atypical antipsychotic agent. It binds several types of central nervous system receptors, and displays a unique pharmacological profile. Clozapine is a serotonin antagonist, with strong binding to 5-HT 2a/2c receptor subtype. It also displays strong affinity to several dopaminergic receptors, but shows only weak antagonism at the dopamine D2 receptor, a receptor commonly thought to modulate neuroleptic activity. Agranulocytosis is a major adverse effect associated with administration of this agent.Second-Generation Antipsychotics
PhenytoinPhenytoinAn anticonvulsant that is used to treat a wide variety of seizures. The mechanism of therapeutic action is not clear, although several cellular actions have been described including effects on ion channels, active transport, and general membrane stabilization. Phenytoin has been proposed for several other therapeutic uses, but its use has been limited by its many adverse effects and interactions with other drugs.First-Generation Anticonvulsant Drugs
Others
CocaineCocaineAn alkaloid ester extracted from the leaves of plants including coca. It is a local anesthetic and vasoconstrictor and is clinically used for that purpose, particularly in the eye, ear, nose, and throat. It also has powerful central nervous system effects similar to the amphetamines and is a drug of abuse. Cocaine, like amphetamines, acts by multiple mechanisms on brain catecholaminergic neurons; the mechanism of its reinforcing effects is thought to involve inhibition of dopamine uptake.Local Anesthetics
Isotretinoin
Epidemiology
IncidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency: approximately 3/100,000
PrevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency: approximately 42/100,000
MPO-AAV is more common than PR3-AAV
Often presents in the 6th and 7th decades of life
Affects women and men equally
More common in White individuals
Drug-induced ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive GlomerulonephritisvasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus:
More common in young females
More commonly associated with skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and Functions damage than other AAVs
Relatively rare involvement of kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy, GI tract, nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification
More commonly associated with pANCA
May be associated with anti-histone antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions
Generally milder disease severity with better prognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
Innate and adaptive immune systemImmune systemThe body’s defense mechanism against foreign organisms or substances and deviant native cells. It includes the humoral immune response and the cell-mediated response and consists of a complex of interrelated cellular, molecular, and genetic components.Primary Lymphatic Organs responses
Intensity and duration of underlying disease process
NeutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation play a central role:
NeutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation have ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis autoantigens sequestered in cytoplasmic granules within the cell (cannot be accessed by serum antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions).
Initiating inflammatory event (e.g., infection, drugs) → cytokine/complement (tumorTumorInflammationnecrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage factor ɑ (TNF-ɑ), IL-1β, C5a)-induced priming of neutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation
Neutrophil priming → MPOMPOAcute Myeloid Leukemia and PR3 move from the cytoplasm to the cell surface → binding to ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive GlomerulonephritisautoantibodiesAutoantibodiesAntibodies that react with self-antigens (autoantigens) of the organism that produced them.Blotting Techniques
Binding of ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive GlomerulonephritisautoantibodiesAutoantibodiesAntibodies that react with self-antigens (autoantigens) of the organism that produced them.Blotting Techniques to MPOMPOAcute Myeloid Leukemia/PR3 → Fcɣ-mediated neutrophil activationNeutrophil activationThe process in which the neutrophil is stimulated by diverse substances, resulting in degranulation and/or generation of reactive oxygen products, and culminating in the destruction of invading pathogens. The stimulatory substances, including opsonized particles, immune complexes, and chemotactic factors, bind to specific cell-surface receptors on the neutrophil.Ehrlichiosis and Anaplasmosis
Neutrophil activationNeutrophil activationThe process in which the neutrophil is stimulated by diverse substances, resulting in degranulation and/or generation of reactive oxygen products, and culminating in the destruction of invading pathogens. The stimulatory substances, including opsonized particles, immune complexes, and chemotactic factors, bind to specific cell-surface receptors on the neutrophil.Ehrlichiosis and Anaplasmosis → complement pathway activation → elaboration of C5a (attracts more neutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation and acts as an additional neutrophil primer)
Activated neutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation adhere to/penetrate vessel walls → acute inflammationAcute InflammationInflammation/generation of reactive oxygen speciesReactive oxygen speciesMolecules or ions formed by the incomplete one-electron reduction of oxygen. These reactive oxygen intermediates include singlet oxygen; superoxides; peroxides; hydroxyl radical; and hypochlorous acid. They contribute to the microbicidal activity of phagocytes, regulation of signal transduction and gene expression, and the oxidative damage to nucleic acids; proteins; and lipids.Metabolic Dysfunction-associated Steatotic Liver Disease (MASLD) → endothelial damage
Acute inflammationAcute InflammationInflammation → activation of fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis/collagenCollagenA polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth).Connective Tissue: Histology deposition (formation of neutrophil extracellular traps (NETs)) → chronic inflammationChronic InflammationInflammation (apoptosisApoptosisA regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth.Ischemic Cell Damage, necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage, fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans)
Clinical Presentation
AAV presents with a wide range of signs and symptoms depending on the organ system(s) involved.
Constitutional symptoms
Constitutional symptoms may be present weeks or months before specific clinical features develop:
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia, lethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia
More common in GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis (95%) than in MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides (35%)
Manifestations include:
SinusitisSinusitisSinusitis refers to inflammation of the mucosal lining of the paranasal sinuses. The condition usually occurs concurrently with inflammation of the nasal mucosa (rhinitis), a condition known as rhinosinusitis. Acute sinusitis is due to an upper respiratory infection caused by a viral, bacterial, or fungal agent. Sinusitis
Otitis media
Purulent/bloody nasal discharge
Mucosal ulcers
Polychondritis
BoneBoneBone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types/cartilageCartilageCartilage is a type of connective tissue derived from embryonic mesenchyme that is responsible for structural support, resilience, and the smoothness of physical actions. Perichondrium (connective tissue membrane surrounding cartilage) compensates for the absence of vasculature in cartilage by providing nutrition and support. Cartilage: Histology destruction:
Loss of teethTeethNormally, an adult has 32 teeth: 16 maxillary and 16 mandibular. These teeth are divided into 4 quadrants with 8 teeth each. Each quadrant consists of 2 incisors (dentes incisivi), 1 canine (dens caninus), 2 premolars (dentes premolares), and 3 molars (dentes molares). Teeth are composed of enamel, dentin, and dental cement.Teeth: Anatomy
Saddle noseNoseThe nose is the human body’s primary organ of smell and functions as part of the upper respiratory system. The nose may be best known for inhaling oxygen and exhaling carbon dioxide, but it also contributes to other important functions, such as tasting. The anatomy of the nose can be divided into the external nose and the nasal cavity. Nose Anatomy (External & Internal)deformityDeformityExamination of the Upper Limbs
Conductive hearing lossConductive hearing lossHearing loss due to interference with the mechanical reception or amplification of sound to the cochlea. The interference is in the outer or middle ear involving the ear canal; tympanic membrane; or ear ossicles.Hearing Loss or saddle noseNoseThe nose is the human body’s primary organ of smell and functions as part of the upper respiratory system. The nose may be best known for inhaling oxygen and exhaling carbon dioxide, but it also contributes to other important functions, such as tasting. The anatomy of the nose can be divided into the external nose and the nasal cavity. Nose Anatomy (External & Internal)deformityDeformityExamination of the Upper Limbs (typical for GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis)
DyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea
WheezingWheezingWheezing is an abnormal breath sound characterized by a whistling noise that can be relatively high-pitched and shrill (more common) or coarse. Wheezing is produced by the movement of air through narrowed or compressed small (intrathoracic) airways. Wheezing
HemoptysisHemoptysisHemoptysis is defined as the expectoration of blood originating in the lower respiratory tract. Hemoptysis is a consequence of another disease process and can be classified as either life threatening or non-life threatening. Hemoptysis can result in significant morbidity and mortality due to both drowning (reduced gas exchange as the lungs fill with blood) and hemorrhagic shock. Hemoptysis
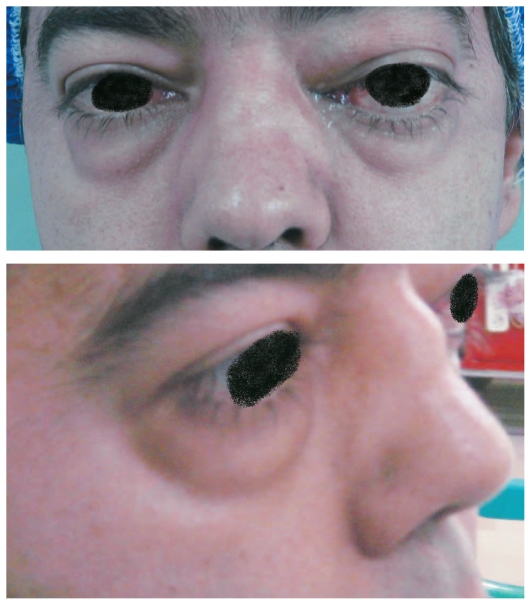
Example of the collapsed nasal bridge (saddle nose deformity) seen in granulomatosis with polyangiitis (GPA): The individual also shows orbital involvement with proptosis (protrusion of the eyeball), eyelid edema, and limited ocular movements. Orbital involvement typically does not occur until years after disease onset.
Image: “Anterior and lateral appearance of the patient” by Aletaha M, Tavakoli M, Kanavi MR, Hashemlou A, Roghaei S. License: CC BY 2.5
Signs of consolidationConsolidationPulmonary Function Tests and/or pleural effusionPleural EffusionPleural effusion refers to the accumulation of fluid between the layers of the parietal and visceral pleura. Common causes of this condition include infection, malignancy, autoimmune disorders, or volume overload. Clinical manifestations include chest pain, cough, and dyspnea. Pleural Effusion, including dullness to percussionPercussionAct of striking a part with short, sharp blows as an aid in diagnosing the condition beneath the sound obtained.Pulmonary Examination
History of asthmaAsthmaAsthma is a chronic inflammatory respiratory condition characterized by bronchial hyperresponsiveness and airflow obstruction. The disease is believed to result from the complex interaction of host and environmental factors that increase disease predisposition, with inflammation causing symptoms and structural changes. Patients typically present with wheezing, cough, and dyspnea. Asthma suggests EGPA over GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis/MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides.
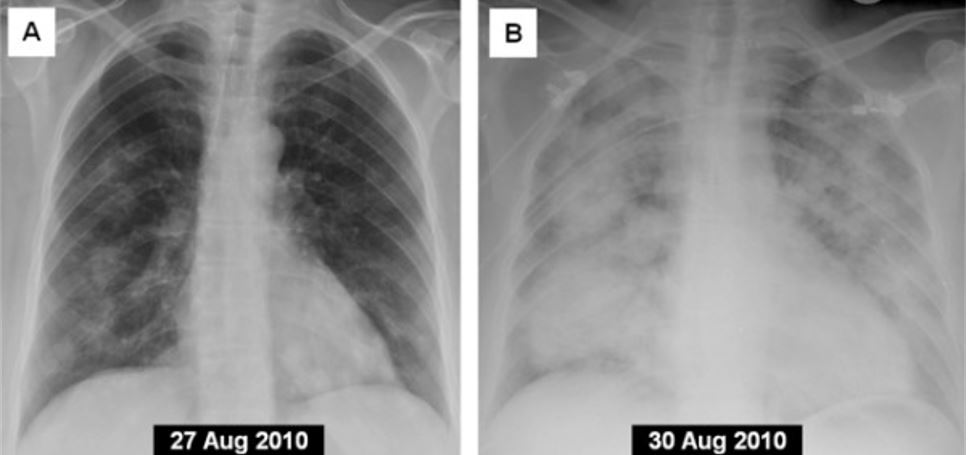
Progression to diffuse alveolar hemorrhage in an individual with granulomatosis with polyangiitis (GPA): A) frontal chest X-ray on admission B) frontal chest X-ray in the same individual after 4 days
Image: “Posterior-anterior chest X-ray 4 days prior to admission” by Cardenas-Garcia J, Farmakiotis D, Baldovino BP, Kim P. License: CC BY 2.0
Kidney involvement
Glomerulonephritis is seen in 20% of individuals at the time of diagnosis and 80% of individuals after 2 years.
HypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension
↑ Creatinine
Rapidly progressive glomerulonephritisRapidly Progressive GlomerulonephritisRapidly progressive glomerulonephritis (RPGN) is a syndrome of severe glomerular disease with progressive loss of kidney function within weeks to months. Histologically, crescents (the proliferation of epithelial cells and the infiltration of monocytes/macrophages in the Bowman space) are found in the glomeruli and arise from immunologic injury.Rapidly Progressive Glomerulonephritis (RPGNRPGNRapidly progressive glomerulonephritis (RPGN) is a syndrome of severe glomerular disease with progressive loss of kidney function within weeks to months. Histologically, crescents (the proliferation of epithelial cells and the infiltration of monocytes/macrophages in the Bowman space) are found in the glomeruli and arise from immunologic injury.Rapidly Progressive Glomerulonephritis): rapidly ↓ renal function over days-to-weeks and glomerular crescentCrescentRapidly Progressive Glomerulonephritis formation on histology
Cutaneous manifestations
Affects 50% of individuals
More commonly observed in drug-associated ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive GlomerulonephritisvasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus than in other forms of AAV
Includes:
Purpura
Ulcerations
UrticariaUrticariaUrticaria is raised, well-circumscribed areas (wheals) of edema (swelling) and erythema (redness) involving the dermis and epidermis with associated pruritus (itch). Urticaria is not a single disease but rather is a reaction pattern representing cutaneous mast cell degranulation.Urticaria (Hives)
Livedo reticularisLivedo reticularisA condition characterized by a reticular or fishnet pattern on the skin of lower extremities and other parts of the body. This red and blue pattern is due to deoxygenated blood in unstable dermal blood vessels. The condition is intensified by cold exposure and relieved by rewarming.Chronic Kidney Disease
Nodules
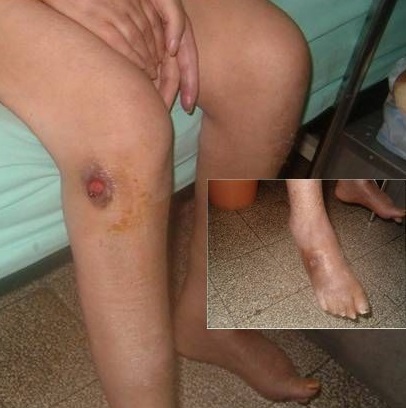
Multiple ulcerations in an individual with microscopic polyangiitis (MPA)
Image: “Ulcérations cutanées aux membres inférieurs” by Khammassi N, Chakroun A. License: CC BY 2.0
Ophthalmic manifestations
PainPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways
Blurred visionBlurred VisionRetinal Detachment/diplopiaDiplopiaA visual symptom in which a single object is perceived by the visual cortex as two objects rather than one. Disorders associated with this condition include refractive errors; strabismus; oculomotor nerve diseases; trochlear nerve diseases; abducens nerve diseases; and diseases of the brain stem and occipital lobe.Myasthenia Gravis
ConjunctivitisConjunctivitisConjunctivitis is a common inflammation of the bulbar and/or palpebral conjunctiva. It can be classified into infectious (mostly viral) and noninfectious conjunctivitis, which includes allergic causes. Patients commonly present with red eyes, increased tearing, burning, foreign body sensation, and photophobia. Conjunctivitis
Episcleritis/scleritisScleritisRefers to any inflammation of the sclera including episcleritis, a benign condition affecting only the episclera, which is generally short-lived and easily treated. Classic scleritis, on the other hand, affects deeper tissue and is characterized by higher rates of visual acuity loss and even mortality, particularly in necrotizing form.Crohn Disease
Corneal ulcerationUlcerationCorneal Abrasions, Erosion, and Ulcers, uveitisUveitisUveitis is the inflammation of the uvea, the pigmented middle layer of the eye, which comprises the iris, ciliary body, and choroid. The condition is categorized based on the site of disease; anterior uveitis is the most common. Diseases of the Uvea, retinitis
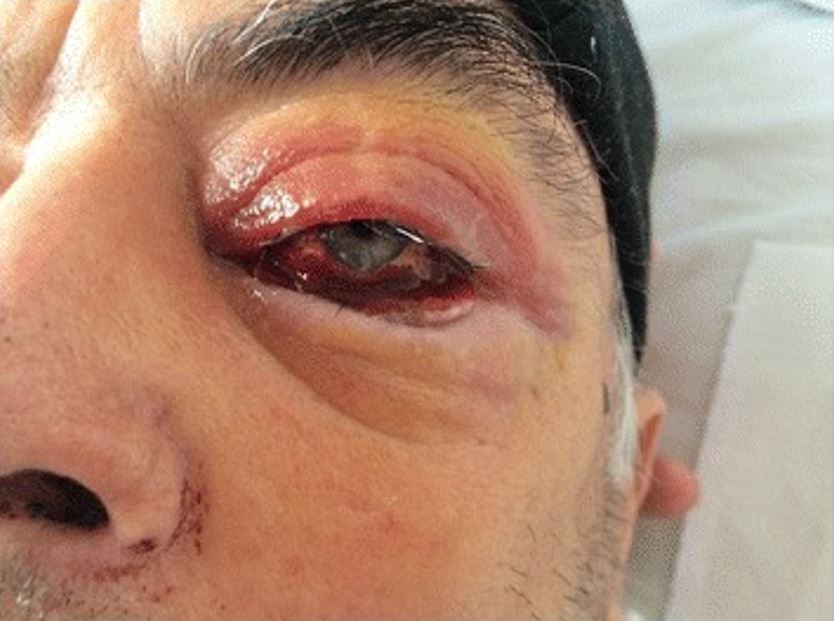
Eyelid edema, scleritis, chemosis, and subconjunctival hemorrhage in an individual with granulomatosis with polyangiitis (GPA)
Image: “The eye involvement of Wegener’s granulomatosis and signs of an acute epistaxis” by Bîrluţiu V, Rezi EC, Bîrluţiu RM, Zaharie IS. License: CC BY 4.0
Sensorineural hearing lossSensorineural hearing lossHearing loss resulting from damage to the cochlea and the sensorineural elements which lie internally beyond the oval and round windows. These elements include the auditory nerve and its connections in the brainstem.Hearing Loss
PericarditisPericarditisPericarditis is an inflammation of the pericardium, often with fluid accumulation. It can be caused by infection (often viral), myocardial infarction, drugs, malignancies, metabolic disorders, autoimmune disorders, or trauma. Acute, subacute, and chronic forms exist. Pericarditis
MyocarditisMyocarditisMyocarditis is an inflammatory disease of the myocardium, which may occur alone or in association with a systemic process. There are numerous etiologies of myocarditis, but all lead to inflammation and myocyte injury, most often leading to signs and symptoms of heart failure. Myocarditis
ThyroidThyroidThe thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck.Thyroid Gland: Anatomy or parathyroidParathyroidThe parathyroid glands are 2 pairs of small endocrine glands found in close proximity to the thyroid gland. The superior parathyroid glands are lodged within the parenchyma of the upper poles of the right and left thyroid lobes; the inferior parathyroid glands are close to the inferior tips or poles of the lobes.Parathyroid Glands: Anatomy disease
Diagnosis
Laboratory evaluation
Serum chemistry:
↑ Creatinine indicates renal involvement.
↓ AlbuminAlbuminSerum albumin from humans. It is an essential carrier of both endogenous substances, such as fatty acids and bilirubin, and of xenobiotics in the blood.Liver Function Tests indicates renal protein losses.
↑ ASTASTEnzymes of the transferase class that catalyze the conversion of l-aspartate and 2-ketoglutarate to oxaloacetate and l-glutamate.Liver Function Tests/ALTALTAn enzyme that catalyzes the conversion of l-alanine and 2-oxoglutarate to pyruvate and l-glutamate.Liver Function Tests indicates hepatic involvement.
Possible electrolyte disturbance due to renal dysfunction
Possible acid–base disorder due to renal dysfunction
UrinalysisUrinalysisExamination of urine by chemical, physical, or microscopic means. Routine urinalysis usually includes performing chemical screening tests, determining specific gravity, observing any unusual color or odor, screening for bacteriuria, and examining the sediment microscopically.Urinary Tract Infections (UTIs) in Children:
↑ IgEIgEAn immunoglobulin associated with mast cells. Overexpression has been associated with allergic hypersensitivity.Immunoglobulins: Types and Functions (in EPGA)
Normocytic normochromic anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Positive immunofluorescence and/or enzyme-linked immunosorbent assay (ELISAELISAAn immunoassay utilizing an antibody labeled with an enzyme marker such as horseradish peroxidase. While either the enzyme or the antibody is bound to an immunosorbent substrate, they both retain their biologic activity; the change in enzyme activity as a result of the enzyme-antibody-antigen reaction is proportional to the concentration of the antigen and can be measured spectrophotometrically or with the naked eye. Many variations of the method have been developed.St. Louis Encephalitis Virus) for ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis (serum):
Proteinase 3Proteinase 3Granulomatosis with Polyangiitis (PR3)-ANCA (c-ANCA): more commonly associated with GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis
MyeloperoxidaseMyeloperoxidaseAcute Myeloid Leukemia (MPOMPOAcute Myeloid Leukemia)-ANCA (p-ANCA): more commonly associated with MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides, EGPA
Imaging
Chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests and CT findings may include:
Pleural effusionPleural EffusionPleural effusion refers to the accumulation of fluid between the layers of the parietal and visceral pleura. Common causes of this condition include infection, malignancy, autoimmune disorders, or volume overload. Clinical manifestations include chest pain, cough, and dyspnea. Pleural Effusion
Fleeting infiltrates
Hilar adenopathy
Diagnostic criteria
American College of Rheumatology diagnosis for EGPA includes 4 of the 6 following criteria with a specificity of almost 100%:
AsthmaAsthmaAsthma is a chronic inflammatory respiratory condition characterized by bronchial hyperresponsiveness and airflow obstruction. The disease is believed to result from the complex interaction of host and environmental factors that increase disease predisposition, with inflammation causing symptoms and structural changes. Patients typically present with wheezing, cough, and dyspnea. Asthma
> 10% eosinophilsEosinophilsGranular leukocytes with a nucleus that usually has two lobes connected by a slender thread of chromatin, and cytoplasm containing coarse, round granules that are uniform in size and stainable by eosin.Innate Immunity: Phagocytes and Antigen Presentation on CBC with differential
MononeuropathyMononeuropathyDisease or trauma involving a single peripheral nerve in isolation, or out of proportion to evidence of diffuse peripheral nerve dysfunction. Mononeuropathy multiplex refers to a condition characterized by multiple isolated nerve injuries. Mononeuropathies may result from a wide variety of causes, including ischemia; traumatic injury; compression; connective tissue diseases; cumulative trauma disorders; and other conditions.Mononeuropathy and Plexopathy (e.g., foot dropFoot DropLeprosy) or polyneuropathyPolyneuropathyPolyneuropathy is any disease process affecting the function of or causing damage to multiple nerves of the peripheral nervous system. There are numerous etiologies of polyneuropathy, most of which are systemic and the most common of which is diabetic neuropathy. Polyneuropathy
Fleeting pulmonary infiltrates
Paranasal sinus abnormality
Extravascular accumulation of eosinophilsEosinophilsGranular leukocytes with a nucleus that usually has two lobes connected by a slender thread of chromatin, and cytoplasm containing coarse, round granules that are uniform in size and stainable by eosin.Innate Immunity: Phagocytes and Antigen Presentation on biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma
BiopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma
Confirmation of diagnosis includes biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma of affected organs:
Suspected diagnosis can be confirmed with a biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma of lung, skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and Functions, or peripheral nerve (may show granulomasGranulomasA relatively small nodular inflammatory lesion containing grouped mononuclear phagocytes, caused by infectious and noninfectious agents.Sarcoidosis).
BiopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma sites and findings:
GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis: Tissue biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma from a site of the active disease shows segmental, crescentic-necrotizing glomerulonephritis with little/no immunoglobulin or complement deposition (pauci-immune); renal and lung biopsies are the most specific.
MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides: Lung biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma and/or renal biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma (crescentic glomerulonephritis) shows necrotizing arteritis (histologically identical to polyarteritis nodosaPolyarteritis nodosaA form of necrotizing non-granulomatous inflammation occurring primarily in medium-sized arteries, often with microaneurysms. It is characterized by muscle, joint, and abdominal pain resulting from arterial infarction and scarring in affected organs. Polyarteritis nodosa with lung involvement is called churg-strauss syndrome.Vasculitides).
EGPA: Lung biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma shows eosinophiliaEosinophiliaAbnormal increase of eosinophils in the blood, tissues or organs.Autosomal Dominant Hyperimmunoglobulin E Syndrome and small, necrotizing eosinophilic granulomasGranulomasA relatively small nodular inflammatory lesion containing grouped mononuclear phagocytes, caused by infectious and noninfectious agents.Sarcoidosis with necrotizing vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus involving small arteriesSmall arteriesArteries: Histology and venulesVenulesThe minute vessels that collect blood from the capillary plexuses and join together to form veins.Veins: Histology
Anti-GBM autoantibody disease: Kidney biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma shows necrotizing glomerulonephritis with little or no deposition of immune complex or complement deposition.
Drug-induced ANCA-associated vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus: No distinguishing pathologic findings for discrimination between drug-induced vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus and other vasculitidesVasculitidesVasculitides are a group of conditions characterized by vasculitis, ischemia, and damage to the organs supplied by the affected vessels. The affected arteries are of different sizes and locations and vary by the type of vasculitis. Vasculitides.
Management
All individuals with a confirmed diagnosis of AAV require immunosuppressive therapy. Therapy can be initiated early based on a presumptive diagnosis of the following criteria:
Suggestive clinical features
+ ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis test
↓ ProbabilityProbabilityProbability is a mathematical tool used to study randomness and provide predictions about the likelihood of something happening. There are several basic rules of probability that can be used to help determine the probability of multiple events happening together, separately, or sequentially.Basics of Probability of other etiologies
Induction of remissionRemissionA spontaneous diminution or abatement of a disease over time, without formal treatment.Cluster Headaches
MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides and GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis:
CyclophosphamideCyclophosphamidePrecursor of an alkylating nitrogen mustard antineoplastic and immunosuppressive agent that must be activated in the liver to form the active aldophosphamide. It has been used in the treatment of lymphoma and leukemia. Its side effect, alopecia, has been used for defleecing sheep. Cyclophosphamide may also cause sterility, birth defects, mutations, and cancer.Immunosuppressants + glucocorticoidsGlucocorticoidsGlucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs.Glucocorticoids are the criterion standard for induction of remissionRemissionA spontaneous diminution or abatement of a disease over time, without formal treatment.Cluster Headaches.
RituximabRituximabA murine-derived monoclonal antibody and antineoplastic agent that binds specifically to the cd20 antigen and is used in the treatment of leukemia; lymphoma and rheumatoid arthritis.Immunosuppressants is a less toxic alternative to cyclophosphamideCyclophosphamidePrecursor of an alkylating nitrogen mustard antineoplastic and immunosuppressive agent that must be activated in the liver to form the active aldophosphamide. It has been used in the treatment of lymphoma and leukemia. Its side effect, alopecia, has been used for defleecing sheep. Cyclophosphamide may also cause sterility, birth defects, mutations, and cancer.Immunosuppressants for induction of remissionRemissionA spontaneous diminution or abatement of a disease over time, without formal treatment.Cluster Headaches in GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis or MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides and is the 1st treatment approved by the FDA.
EGPA:
GlucocorticoidsGlucocorticoidsGlucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs.Glucocorticoids alone are usually adequate for the treatment of EGPA.
CytotoxicCytotoxicParvovirus B19 drugs (cyclophosphamideCyclophosphamidePrecursor of an alkylating nitrogen mustard antineoplastic and immunosuppressive agent that must be activated in the liver to form the active aldophosphamide. It has been used in the treatment of lymphoma and leukemia. Its side effect, alopecia, has been used for defleecing sheep. Cyclophosphamide may also cause sterility, birth defects, mutations, and cancer.Immunosuppressants) are necessary in < 20% of individuals.
Plasma exchangePlasma exchangeRemoval of plasma and replacement with various fluids, e.g., fresh frozen plasma, plasma protein fractions (ppf), albumin preparations, dextran solutions, saline. Used in treatment of autoimmune diseases, immune complex diseases, diseases of excess plasma factors, and other conditions.Thrombotic Thrombocytopenic Purpura may be required with disease (e.g., RPGNRPGNRapidly progressive glomerulonephritis (RPGN) is a syndrome of severe glomerular disease with progressive loss of kidney function within weeks to months. Histologically, crescents (the proliferation of epithelial cells and the infiltration of monocytes/macrophages in the Bowman space) are found in the glomeruli and arise from immunologic injury.Rapidly Progressive Glomerulonephritis or pulmonary hemorrhage).
Maintenance of remissionRemissionA spontaneous diminution or abatement of a disease over time, without formal treatment.Cluster Headaches
Once remissionRemissionA spontaneous diminution or abatement of a disease over time, without formal treatment.Cluster Headaches is achieved, individuals switch to less toxic agents:
AzathioprineAzathioprineAn immunosuppressive agent used in combination with cyclophosphamide and hydroxychloroquine in the treatment of rheumatoid arthritis. According to the fourth annual report on carcinogens, this substance has been listed as a known carcinogen.Immunosuppressants
RituximabRituximabA murine-derived monoclonal antibody and antineoplastic agent that binds specifically to the cd20 antigen and is used in the treatment of leukemia; lymphoma and rheumatoid arthritis.Immunosuppressants
MethotrexateMethotrexateAn antineoplastic antimetabolite with immunosuppressant properties. It is an inhibitor of tetrahydrofolate dehydrogenase and prevents the formation of tetrahydrofolate, necessary for synthesis of thymidylate, an essential component of DNA.Antimetabolite Chemotherapy
Trimethoprim-sulfamethoxazole to prevent pneumocystis pneumoniaPneumoniaPneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy.Pneumonia (PCPPCPPneumocystis jiroveci is a yeast-like fungus causing pneumocystis pneumonia (PCP) in immunocompromised patients. Pneumocystis pneumonia is spread through airborne transmission and classically affects patients with AIDS, functioning as an AIDS-defining illness. Patients may present with insidious onset of fever, chills, dry cough, chest pain, and shortness of breath.Pneumocystis jirovecii/Pneumocystis Pneumonia (PCP))
Comparison Table
Table showing suggestive laboratory and clinical features of the 3 main types of AAV:
Table: Suggestive laboratory and clinical features of the 3 main types of AAV
AAV disease
ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis
Granuloma on biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma
Typical case
GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis
MPAMPAA primary systemic vasculitis of small- and some medium-sized vessels. It is characterized by a tropism for kidneys and lungs, positive association with anti-neutrophil cytoplasmic antibodies (ANCA), and a paucity of immunoglobulin deposits in vessel walls.Vasculitides
In 70%
p-ANCA
–
Similar to GPAGPAA multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin.Granulomatosis with Polyangiitis, but without serious nasal/sinus disease (only in 30%)
Cutaneous involvementCutaneous involvementCoccidioides/Coccidioidomycosis with a history of asthmaAsthmaAsthma is a chronic inflammatory respiratory condition characterized by bronchial hyperresponsiveness and airflow obstruction. The disease is believed to result from the complex interaction of host and environmental factors that increase disease predisposition, with inflammation causing symptoms and structural changes. Patients typically present with wheezing, cough, and dyspnea. Asthma/allergic rhinitisAllergic rhinitisAn inflammation of the nasal mucosa triggered by allergens.Rhinitis + eosinophiliaEosinophiliaAbnormal increase of eosinophils in the blood, tissues or organs.Autosomal Dominant Hyperimmunoglobulin E Syndrome
GPA: granulomatosis with polyangiitis MPA: microscopic polyangiitis EGPA: eosinophilic granulomatosis with polyangiitis
Differential Diagnosis
Drug-induced ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive GlomerulonephritisvasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus:ANCAANCAGroup of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls.Rapidly Progressive Glomerulonephritis positive vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus from medication exposures are linked to the development of vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus (propylthiouracilPropylthiouracilA thiourea antithyroid agent. Propylthiouracil inhibits the synthesis of thyroxine and inhibits the peripheral conversion of thyroxine to triiodothyronine. It is used in the treatment of hyperthyroidism.Antithyroid Drugs, methimazoleMethimazoleA thioureylene antithyroid agent that inhibits the formation of thyroid hormones by interfering with the incorporation of iodine into tyrosyl residues of thyroglobulin. This is done by interfering with the oxidation of iodide ion and iodotyrosyl groups through inhibition of the peroxidase enzyme.Antithyroid Drugs, carbimazole, hydralazineHydralazineA direct-acting vasodilator that is used as an antihypertensive agent.Heart Failure and Chronic Coronary Syndrome Medication, and minocyclineMinocyclineA tetracycline analog, having a 7-dimethylamino and lacking the 5 methyl and hydroxyl groups, which is effective against tetracycline-resistant staphylococcus infections.Tetracyclines), causing constitutional symptoms such as arthralgias, fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia, and skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and FunctionsrashRashRocky Mountain Spotted Fever. However, the full range of clinical features (including rapidly progressive renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome and alveolar hemorrhage) can also occur.
Goodpasture syndromeGoodpasture SyndromeGoodpasture syndrome, also known as anti-glomerular basement membrane (GBM) disease, is an autoimmune disease characterized by circulating antibodies directed against glomerular and alveolar basement membranes. Affected individuals present with symptoms of rapidly progressive glomerulonephritis and alveolar hemorrhage. Goodpasture Syndrome: a rare autoimmune disease where antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions attack the basement membraneBasement membraneA darkly stained mat-like extracellular matrix (ecm) that separates cell layers, such as epithelium from endothelium or a layer of connective tissue. The ecm layer that supports an overlying epithelium or endothelium is called basal lamina. Basement membrane (bm) can be formed by the fusion of either two adjacent basal laminae or a basal lamina with an adjacent reticular lamina of connective tissue. Bm, composed mainly of type IV collagen; glycoprotein laminin; and proteoglycan, provides barriers as well as channels between interacting cell layers.Thin Basement Membrane Nephropathy (TBMN) in the lungsLungsLungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy and kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy, which leads to bleeding from the lungsLungsLungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy and kidney failure. Goodpasture syndromeGoodpasture SyndromeGoodpasture syndrome, also known as anti-glomerular basement membrane (GBM) disease, is an autoimmune disease characterized by circulating antibodies directed against glomerular and alveolar basement membranes. Affected individuals present with symptoms of rapidly progressive glomerulonephritis and alveolar hemorrhage. Goodpasture Syndrome results in permanent lung and kidney damage and often leads to death. Treatment includes immune systemImmune systemThe body’s defense mechanism against foreign organisms or substances and deviant native cells. It includes the humoral immune response and the cell-mediated response and consists of a complex of interrelated cellular, molecular, and genetic components.Primary Lymphatic Organs suppressing medications (e.g., corticosteroidsCorticosteroidsChorioretinitis and cyclophosphamideCyclophosphamidePrecursor of an alkylating nitrogen mustard antineoplastic and immunosuppressive agent that must be activated in the liver to form the active aldophosphamide. It has been used in the treatment of lymphoma and leukemia. Its side effect, alopecia, has been used for defleecing sheep. Cyclophosphamide may also cause sterility, birth defects, mutations, and cancer.Immunosuppressants) and plasmapheresisPlasmapheresisProcedure whereby plasma is separated and extracted from anticoagulated whole blood and the red cells retransfused to the donor. Plasmapheresis is also employed for therapeutic use.Stevens-Johnson Syndrome.
Polyarteritis nodosaPolyarteritis nodosaA form of necrotizing non-granulomatous inflammation occurring primarily in medium-sized arteries, often with microaneurysms. It is characterized by muscle, joint, and abdominal pain resulting from arterial infarction and scarring in affected organs. Polyarteritis nodosa with lung involvement is called churg-strauss syndrome.Vasculitides: a systemic vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus of small and medium vessels. Polyarteritis nodosaPolyarteritis nodosaA form of necrotizing non-granulomatous inflammation occurring primarily in medium-sized arteries, often with microaneurysms. It is characterized by muscle, joint, and abdominal pain resulting from arterial infarction and scarring in affected organs. Polyarteritis nodosa with lung involvement is called churg-strauss syndrome.Vasculitides involves the skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and Functions, peripheral nervesPeripheral NervesThe nerves outside of the brain and spinal cord, including the autonomic, cranial, and spinal nerves. Peripheral nerves contain non-neuronal cells and connective tissue as well as axons. The connective tissue layers include, from the outside to the inside, the epineurium, the perineurium, and the endoneurium.Nervous System: Histology, muscles, joints, gastrointestinal tract, and kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy. Individuals present with nonspecific symptoms, hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, risk of myocardial infarctionMyocardial infarctionMI is ischemia and death of an area of myocardial tissue due to insufficient blood flow and oxygenation, usually from thrombus formation on a ruptured atherosclerotic plaque in the epicardial arteries. Clinical presentation is most commonly with chest pain, but women and patients with diabetes may have atypical symptoms.Myocardial Infarction, and polyneuropathyPolyneuropathyPolyneuropathy is any disease process affecting the function of or causing damage to multiple nerves of the peripheral nervous system. There are numerous etiologies of polyneuropathy, most of which are systemic and the most common of which is diabetic neuropathy. Polyneuropathy. AngiographyAngiographyRadiography of blood vessels after injection of a contrast medium.Cardiac Surgery shows a beading appearance of alternating aneurysms and vessel stenosisStenosisHypoplastic Left Heart Syndrome (HLHS). Management is immunosuppressive regimens.
Giant cell arteritisGiant Cell ArteritisGiant cell arteritis (GCA), also known as temporal arteritis, is a type of large-vessel vasculitis that predominantly affects the aorta and its major branches, with a predilection for the branches of the carotid (including the temporal artery). Giant cell arteritis is defined by inflammatory leukocytes in the vessel walls leading to reactive damage, ischemia, and necrosis. Giant Cell Arteritis: a vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus of medium and large arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology (particularly the carotid arteriesCarotid ArteriesEither of the two principal arteries on both sides of the neck that supply blood to the head and neck; each divides into two branches, the internal carotid artery and the external carotid artery.Carotid Arterial System: Anatomy and aortaAortaThe main trunk of the systemic arteries.Mediastinum and Great Vessels: Anatomy). Individuals present with new-onset headacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess, tender/hardened temporal artery, jaw claudicationJaw claudicationMandibular pain or fatigue during mastication that is relieved with restGiant Cell Arteritis, and amaurosis fugaxAmaurosis fugaxTransient complete or partial monocular blindness due to retinal ischemia. This may be caused by emboli from the carotid artery (usually in association with carotid stenosis) and other locations that enter the central retinal artery.Carotid Artery Stenosis. Laboratory studies show elevated ESRESRSoft Tissue Abscess and CRP. BiopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma shows mononuclear infiltration of vessel walls and the formation of giant cellsGiant cellsMultinucleated masses produced by the fusion of many cells; often associated with viral infections. In aids, they are induced when the envelope glycoprotein of the HIV virus binds to the CD4 antigen of uninfected neighboring T4 cells. The resulting syncytium leads to cell death and thus may account for the cytopathic effect of the virus.Giant Cell Arteritis. Treatment involves prompt administration of glucocorticoidsGlucocorticoidsGlucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs.Glucocorticoids.
Henoch-Schönlein purpuraHenoch-Schönlein PurpuraHenoch-Schönlein purpura (HSP), also known as immunoglobulin A vasculitis, is an autoimmune small-vessel vasculitis that typically presents as a tetrad of abdominal pain, arthralgia, hematuria, and purpuric rash.Henoch-Schönlein Purpura: an autoimmune small vessel vasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus. The condition typically presents as a triad of abdominal painAbdominal PainAcute Abdomen, hematuriaHematuriaPresence of blood in the urine.Renal Cell Carcinoma, and purpuric rashRashRocky Mountain Spotted Fever. The pathophysiology involves IgAIgARepresents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions.Immunoglobulins: Types and Functions immune complex deposition in multiple vessels following a triggerTriggerThe type of signal that initiates the inspiratory phase by the ventilatorInvasive Mechanical Ventilation. Symptoms depend on the tissues supplied by the vessels. The disease has a clinical diagnosis and is managed symptomatically.
Rheumatoid arthritisArthritisAcute or chronic inflammation of joints.Osteoarthritis: an inflammatory polyarthritisPolyarthritisRheumatoid Arthritis presenting with painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways in symmetric joints. The painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways is worse in the morning but improves during the day, The proximal interphalangeal and metacarpophalangeal jointsMetacarpophalangeal jointsThe articulation between a metacarpal bone and a phalanx.Examination of the Upper Limbs are included, but the distal interphalangeal jointsInterphalangeal jointsHand: Anatomy are spared. Management is with NSAIDsNSAIDSPrimary vs Secondary Headaches, methotrexateMethotrexateAn antineoplastic antimetabolite with immunosuppressant properties. It is an inhibitor of tetrahydrofolate dehydrogenase and prevents the formation of tetrahydrofolate, necessary for synthesis of thymidylate, an essential component of DNA.Antimetabolite Chemotherapy, sulfasalazineSulfasalazineA drug that is used in the management of inflammatory bowel diseases. Its activity is generally considered to lie in its metabolic breakdown product, 5-aminosalicylic acid released in the colon.Sulfonamides and Trimethoprim, hydroxychloroquineHydroxychloroquineA chemotherapeutic agent that acts against erythrocytic forms of malarial parasites. Hydroxychloroquine appears to concentrate in food vacuoles of affected protozoa. It inhibits plasmodial heme polymerase.Immunosuppressants, and TNF-α inhibitors.
Systemic lupus erythematosusSystemic lupus erythematosusSystemic lupus erythematosus (SLE) is a chronic autoimmune, inflammatory condition that causes immune-complex deposition in organs, resulting in systemic manifestations. Women, particularly those of African American descent, are more commonly affected.Systemic Lupus Erythematosus: a chronic inflammatory condition with involvement of the skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and Functions, joints, kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy, blood cells, and CNS. The condition is an autoimmune disorderAutoimmune DisorderSeptic Arthritis associated with the formation of autoantibodiesAutoantibodiesAntibodies that react with self-antigens (autoantigens) of the organism that produced them.Blotting Techniques such as ANA, anti-Smith, and anti-double-stranded DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure (dsDNA). Features include malar rashMalar RashSystemic Lupus Erythematosus, joint painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways, feverFeverFever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever, proteinuriaProteinuriaThe presence of proteins in the urine, an indicator of kidney diseases.Nephrotic Syndrome in Children, hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types, lymphopenia, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, and/or psychosis.
Aratani, Y. (2018). Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 640:47-52. https://pubmed.ncbi.nlm.nih.gov/29336940/