Thrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital Congenital Chorioretinitis or an acquired deficiency of ADAMTS-13, a metalloproteinase that cleaves multimers of von Willebrand factor von Willebrand factor A high-molecular-weight plasma protein, produced by endothelial cells and megakaryocytes, that is part of the factor VIII/von Willebrand factor complex. The von Willebrand factor has receptors for collagen, platelets, and ristocetin activity as well as the immunologically distinct antigenic determinants. It functions in adhesion of platelets to collagen and hemostatic plug formation. The prolonged bleeding time in von Willebrand diseases is due to the deficiency of this factor. Hemostasis (VWF). The large multimers then aggregate excessive platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology resulting in microvascular thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus and an increase in consumption of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology. Clinical presentation can consist of thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia, hematuria Hematuria Presence of blood in the urine. Renal Cell Carcinoma, gastrointestinal symptoms, neurological symptoms, and renal involvement. Diagnosis is established based on a combination of clinical symptoms and laboratory tests. Thrombotic thrombocytopenic purpura is a medical emergency and almost always fatal if appropriate treatment is not initiated promptly. Emergency management includes plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products exchange and immunosuppressive therapies.
Last updated: Dec 15, 2025
Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy caused by deficiency of ADAMTS-13, a von Willebrand factor von Willebrand factor A high-molecular-weight plasma protein, produced by endothelial cells and megakaryocytes, that is part of the factor VIII/von Willebrand factor complex. The von Willebrand factor has receptors for collagen, platelets, and ristocetin activity as well as the immunologically distinct antigenic determinants. It functions in adhesion of platelets to collagen and hemostatic plug formation. The prolonged bleeding time in von Willebrand diseases is due to the deficiency of this factor. Hemostasis (VWF) cleaving protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS.
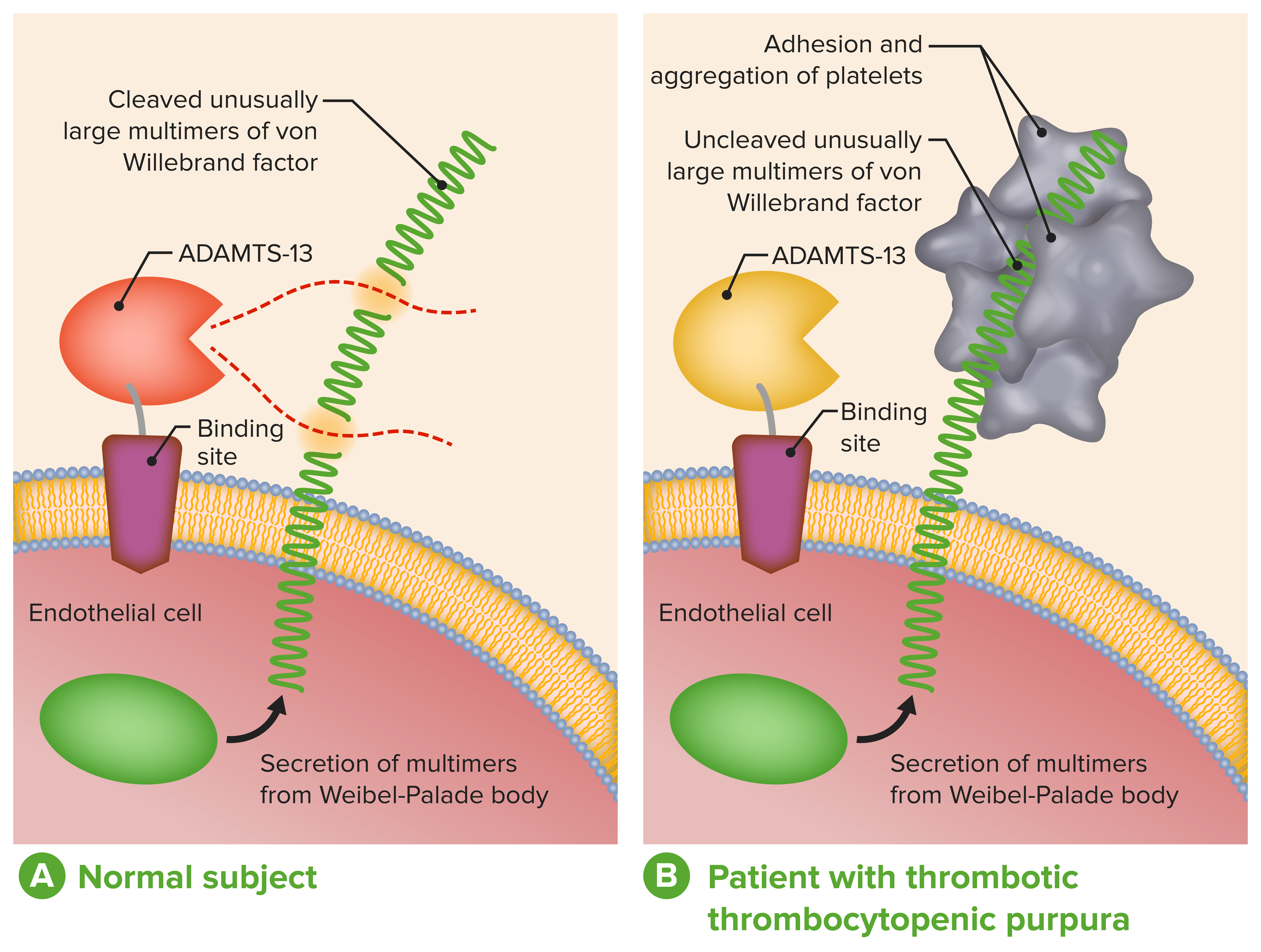
ADAMTS-13 in the diagnosis and management of thrombotic microangiopathies:
A: Normal physiological function of ADAMTS-13
B: Absent or severely reduced activity of ADAMTS-13 in patients with TTP prevents timely cleavage of unusually large multimers of VWF during secretion by endothelial cells.
Classic TTP pentad:
Organ-system manifestations:
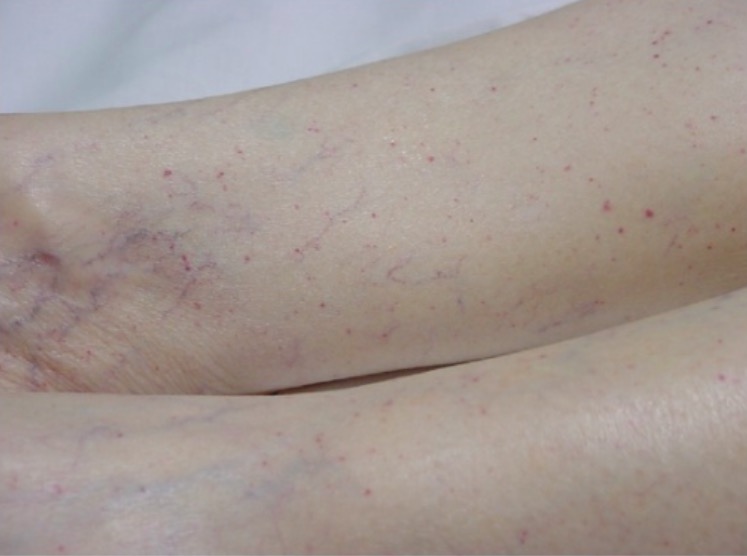
Typical petechial rash on a patient’s lower extremities
Image: “Typical petechial rash” by Department of Emergency Medicine, University of Florida, Gainesville, FL 32610, USA. License: CC BY 3.0Unlike acquired TTP, congenital Congenital Chorioretinitis TTP does not present with acute episodes.
Symptoms are usually mild, vague, and continuous:
Severe and life-threatening exacerbations are most likely during 2 life periods:
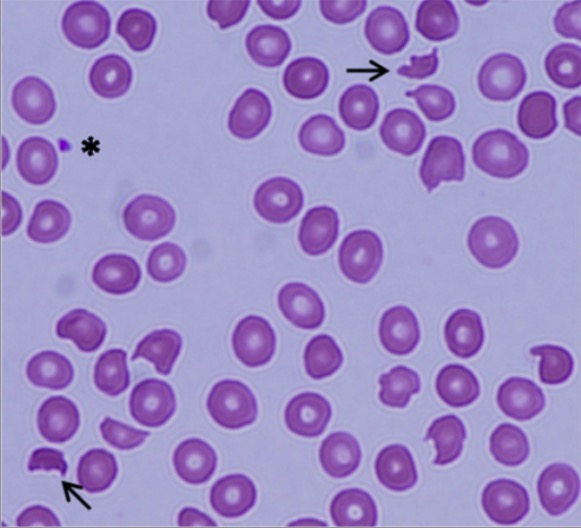
Thrombotic thrombocytopenic purpura (TTP):
Peripheral blood smear showing schistocytes (arrows) and a marked decrease in platelet numbers, indicating microangiopathic hemolytic anemia
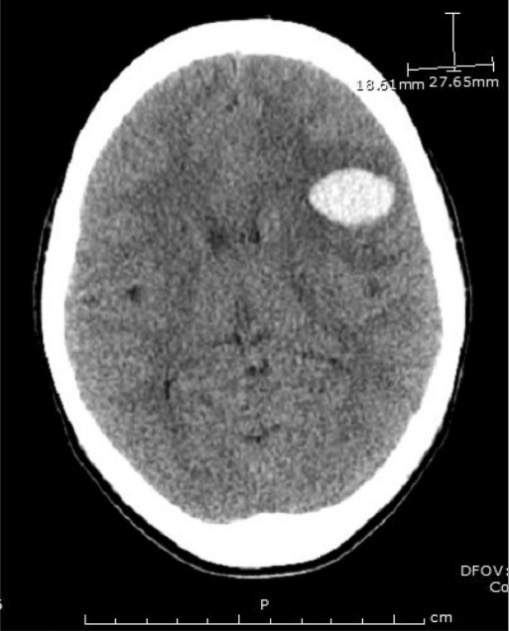
Head CT in a patient demonstrating the larger of 2 frontoparietal hemorrhages
Image: “Emergent head CT demonstrating the larger of two frontoparietal hemorrhages” by Divisions of Infectious Disease, The George Washington University, School of Medicine and Health Sciences, Washington, DC, USA. License: CC BY 3.0The PLASMIC score predicts the probability Probability Probability is a mathematical tool used to study randomness and provide predictions about the likelihood of something happening. There are several basic rules of probability that can be used to help determine the probability of multiple events happening together, separately, or sequentially. Basics of Probability of TTP in the presence of schistocytes Schistocytes Hemolytic Uremic Syndrome on blood smear Blood smear Myeloperoxidase Deficiency.
PLASMIC criteria (1 point for each):
Score: