Dermatomyositis Dermatomyositis A subacute or chronic inflammatory disease of muscle and skin, marked by proximal muscle weakness and a characteristic skin rash. The illness occurs with approximately equal frequency in children and adults. The skin lesions usually take the form of a purplish rash (or less often an exfoliative dermatitis) involving the nose, cheeks, forehead, upper trunk, and arms. The disease is associated with a complement mediated intramuscular microangiopathy, leading to loss of capillaries, muscle ischemia, muscle-fiber necrosis, and perifascicular atrophy. The childhood form of this disease tends to evolve into a systemic vasculitis. Dermatomyositis may occur in association with malignant neoplasms. Paraneoplastic Syndromes ( DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus) is an autoimmune and inflammatory myopathy. Although the etiology of DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus is unclear, it has several genetic and environmental associations. Dermatomyositis Dermatomyositis A subacute or chronic inflammatory disease of muscle and skin, marked by proximal muscle weakness and a characteristic skin rash. The illness occurs with approximately equal frequency in children and adults. The skin lesions usually take the form of a purplish rash (or less often an exfoliative dermatitis) involving the nose, cheeks, forehead, upper trunk, and arms. The disease is associated with a complement mediated intramuscular microangiopathy, leading to loss of capillaries, muscle ischemia, muscle-fiber necrosis, and perifascicular atrophy. The childhood form of this disease tends to evolve into a systemic vasculitis. Dermatomyositis may occur in association with malignant neoplasms. Paraneoplastic Syndromes is common in women around the age of 50 years. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with symmetrical Symmetrical Dermatologic Examination, proximal weakness, characteristic skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions manifestations, and systemic symptoms. Diagnosis is based on clinical presentation and laboratory studies and confirmed on the basis of muscle biopsy Muscle Biopsy Trichinella/Trichinellosis. Myositis-specific antibodies Antibodies Immunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution. Immunoglobulins: Types and Functions, including anti-Mi-2, are specific markers in DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus. Management is with systemic glucocorticoids Systemic Glucocorticoids Glucocorticoids, immunosuppressants Immunosuppressants Immunosuppressants are a class of drugs widely used in the management of autoimmune conditions and organ transplant rejection. The general effect is dampening of the immune response. Immunosuppressants, and physiotherapy Physiotherapy Spinal Stenosis. As there is a strong association of DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus with malignancy Malignancy Hemothorax, all patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship should undergo cancer screening Screening Preoperative Care.
Last updated: Dec 15, 2025
Dermatomyositis Dermatomyositis A subacute or chronic inflammatory disease of muscle and skin, marked by proximal muscle weakness and a characteristic skin rash. The illness occurs with approximately equal frequency in children and adults. The skin lesions usually take the form of a purplish rash (or less often an exfoliative dermatitis) involving the nose, cheeks, forehead, upper trunk, and arms. The disease is associated with a complement mediated intramuscular microangiopathy, leading to loss of capillaries, muscle ischemia, muscle-fiber necrosis, and perifascicular atrophy. The childhood form of this disease tends to evolve into a systemic vasculitis. Dermatomyositis may occur in association with malignant neoplasms. Paraneoplastic Syndromes ( DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus) is an idiopathic, immune-mediated, inflammatory myopathy that causes progressive, symmetric, proximal muscle weakness Proximal Muscle Weakness Lambert-Eaton Myasthenic Syndrome and characteristic cutaneous manifestations.
Antisynthetase syndrome is a subtype of DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus characterized by the presence of antisynthetase antibodies Antibodies Immunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution. Immunoglobulins: Types and Functions and accompanied by certain extramuscular manifestations.
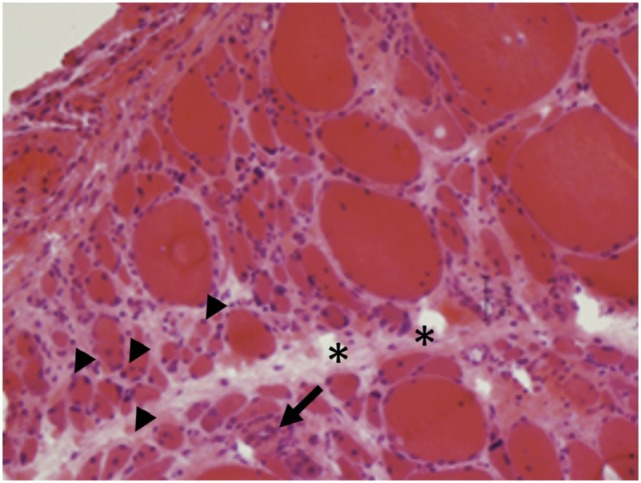
Histological findings of dermatomyositis:
Perifascicular atrophy (arrowheads) and inflammatory infiltrates (arrows) are characteristics of dermatomyositis.
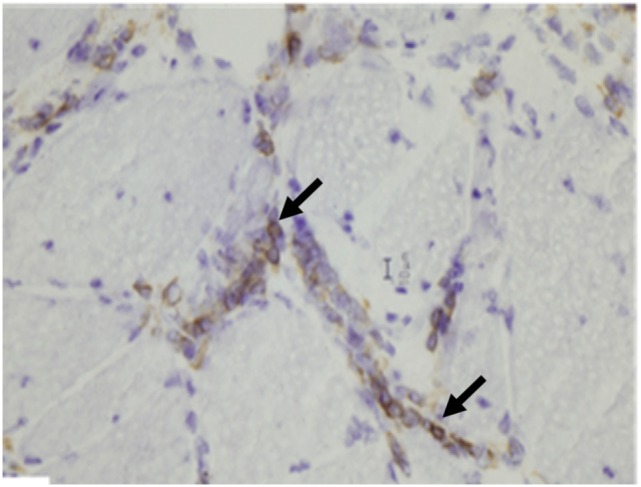
Histological findings of dermatomyositis:
Aggregates of B lymphocytes (arrows) positive for CD20 immunohistochemical stain are found in dermatomyositis.
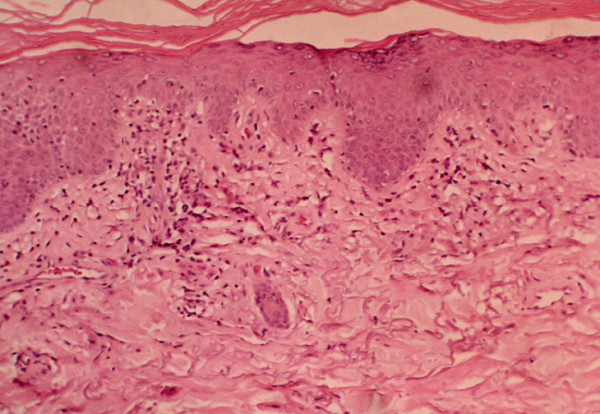
Histological findings of dermatomyositis:
Vacuolar changes of columnar epithelium and lymphocytic inflammatory infiltrate at the dermal-epidermal interface are found in dermatomyositis.
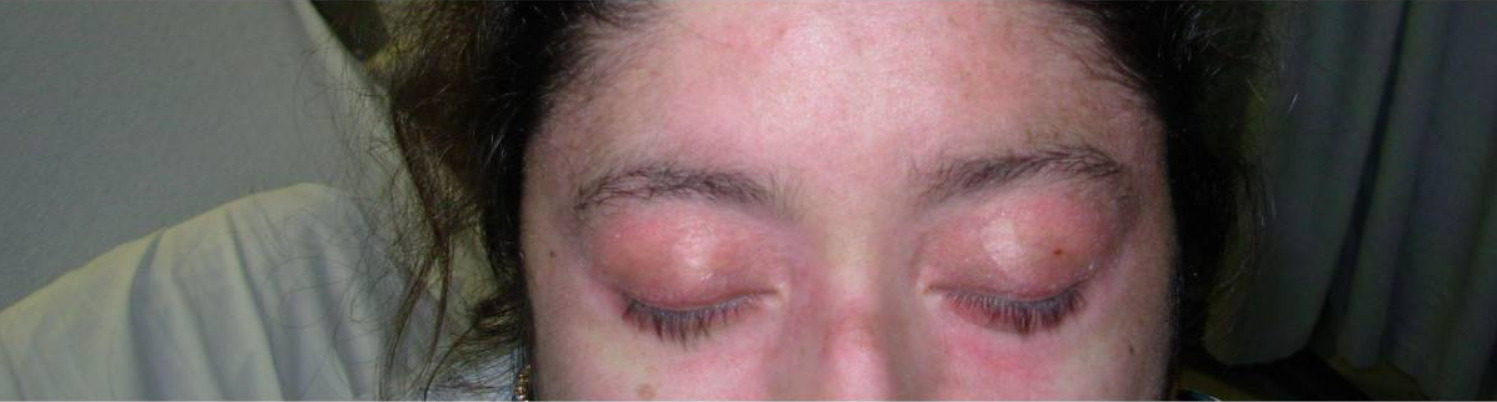
Heliotrope rash in dermatomyositis:
An erythematous rash can be seen around the upper eyelids with periorbital edema.
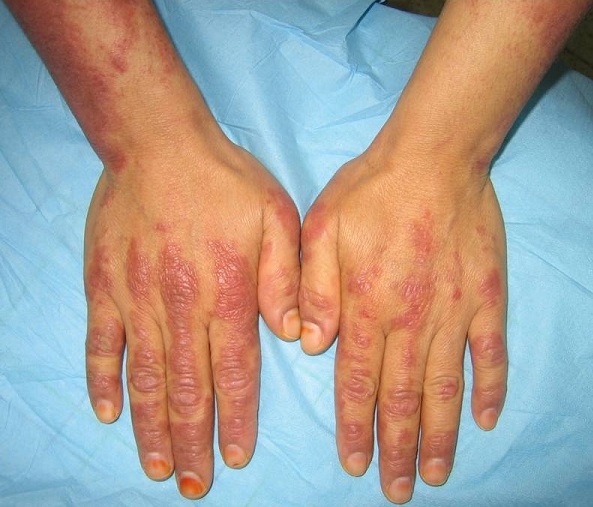
Gottron’s papules in dermatomyositis:
A cutaneous manifestation is seen on the dorsal metacarpophalangeal and interphalangeal joints.
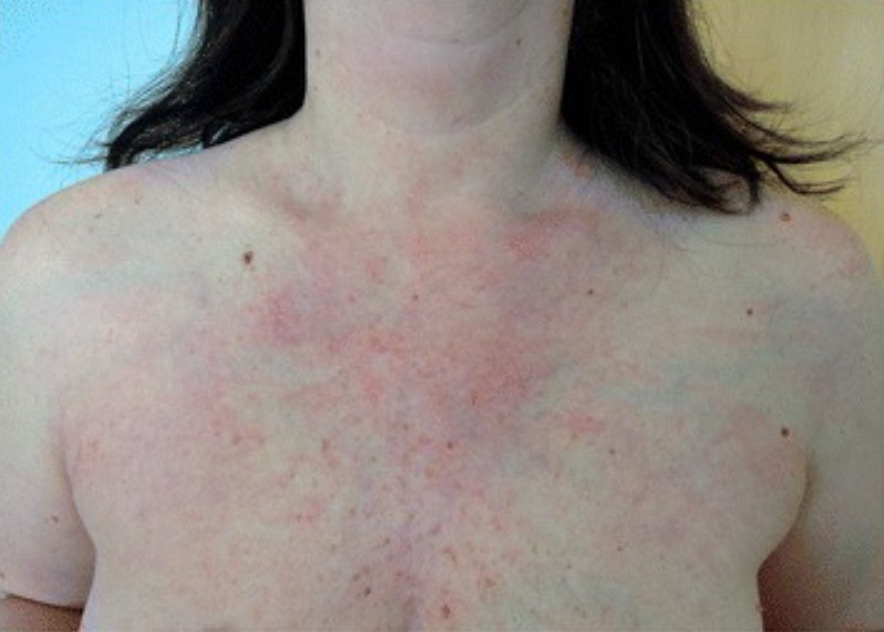
V-sign seen in dermatomyositis: a macular, photo-distributed, erythematous rash resembling the letter V on the anterior aspect of the chest
Image: “Dermatomyositis with anti-TIF-1γ antibodies as a presenting symptom of underlying triple-negative breast cancer: A case report” by BMC Cancer. License: CC BY 4.0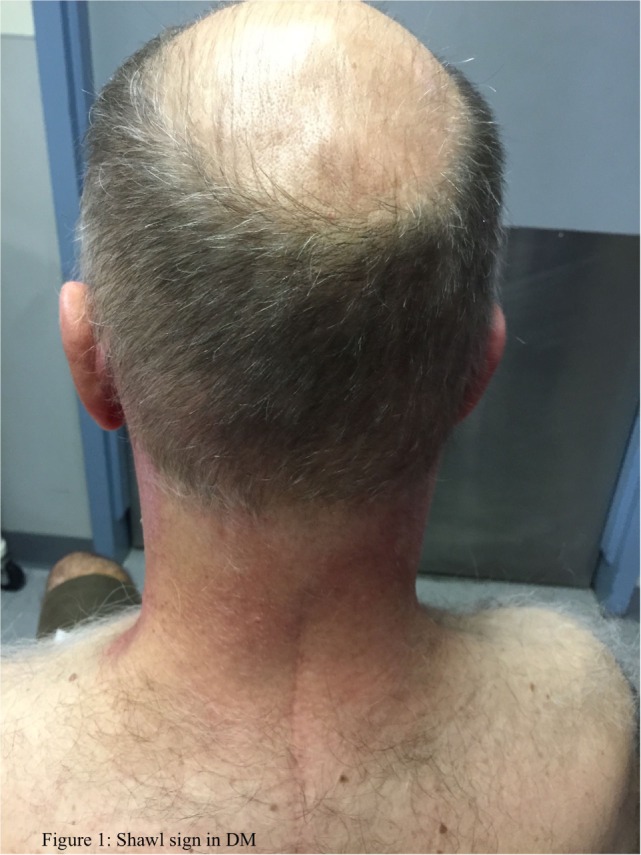
Shawl sign in dermatomyositis
Image: “Idiopathic inflammatory myopathies: Clinical approach and management” by Malik A, Hayat G, Kalia JS, Guzman MA. License: CC BY 4.0Management of DM DM Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus is directed toward the restoration of muscle strength Muscle strength The amount of force generated by muscle contraction. Muscle strength can be measured during isometric, isotonic, or isokinetic contraction, either manually or using a device such as a muscle strength dynamometer. Neurological Examination and minimization of inflammation Inflammation Inflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation.