Becker muscular dystrophy (BMD) is an X-linked recessive X-Linked Recessive Duchenne Muscular Dystrophy genetic disorder that is caused by a mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics. Abnormal, partially functional muscle dystrophin Dystrophin A muscle protein localized in surface membranes which is the product of the Duchenne/Becker muscular dystrophy gene. Individuals with Duchenne muscular dystrophy usually lack dystrophin completely while those with Becker muscular dystrophy have dystrophin of an altered size. It shares features with other cytoskeletal proteins such as spectrin and alpha-actinin but the precise function of dystrophin is not clear. One possible role might be to preserve the integrity and alignment of the plasma membrane to the myofibrils during muscle contraction and relaxation. Blotting Techniques protein is produced, which leads to progressive muscle weakness and the eventual loss of ambulation. The clinical course is highly variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables, but symptoms generally occur by adolescence. The diagnosis is based on muscle enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes, genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies, and muscle biopsy Muscle Biopsy Trichinella/Trichinellosis (if necessary). Management of BMD is supportive and aimed at slowing disease progression and complications. Dilated cardiomyopathy Dilated Cardiomyopathy Dilated cardiomyopathy (DCM) is the most common type of non-ischemic cardiomyopathy and a common cause of heart failure (HF). The cause may be idiopathic, familial, or secondary to a variety of underlying conditions. The disease is characterized by the enlargement of 1 or both ventricles and reduced systolic function. Dilated Cardiomyopathy is the leading cause of death.
Last updated: Dec 15, 2025
Becker muscular dystrophy (BMD) is the 2nd most common form of muscular dystrophy.
Becker muscular dystrophy is caused by non-frameshift deletion or duplication mutations of the DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics on the short arm Arm The arm, or “upper arm” in common usage, is the region of the upper limb that extends from the shoulder to the elbow joint and connects inferiorly to the forearm through the cubital fossa. It is divided into 2 fascial compartments (anterior and posterior). Arm: Anatomy of the X chromosome X chromosome The female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species. Basic Terms of Genetics.
Normal physiology:
Pathophysiology in BMD:
Becker muscular dystrophy has a highly variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables presentation.
Becker muscular dystrophy follows a course of slowly progressive muscle weakness in a proximal-to-distal pattern.
Symptoms:
Physical exam findings:
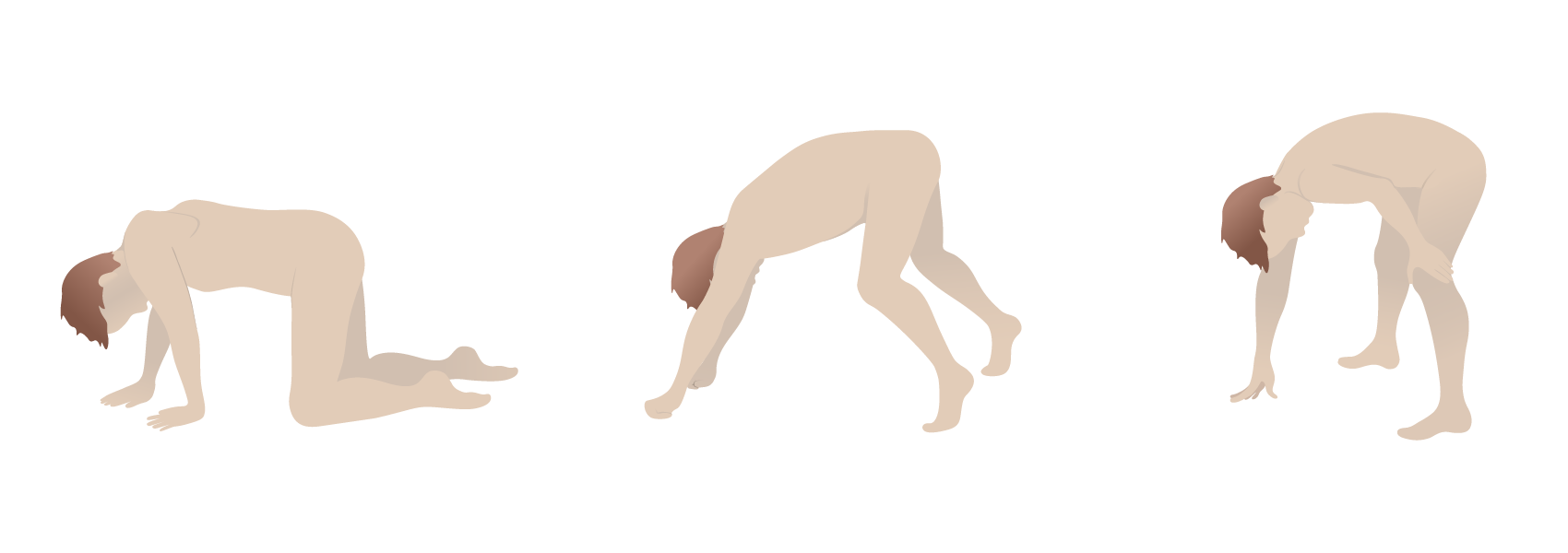
Gower’s sign: the use of the arms and hands to maneuver up to a standing position due to proximal muscle weakness
Image by Lecturio.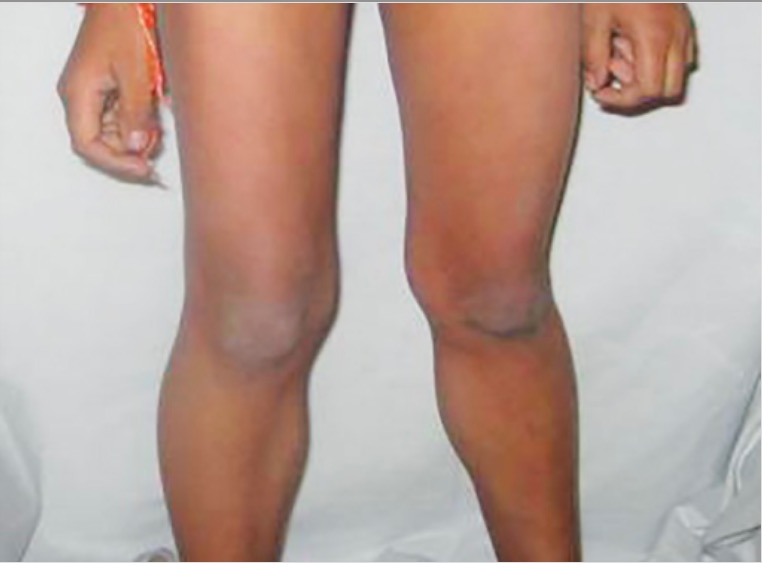
Calf pseudohypertrophy due to muscular dystrophy: large, weak gastrocnemius muscles due to replacement with fat and fibrous tissue
Image: “Calf hypertrophy” by Professor, Department of Pedodontics and Preventive Dentistry, BJS Dental College, Ludhiana, Punjab, India. License: CC BY 3.0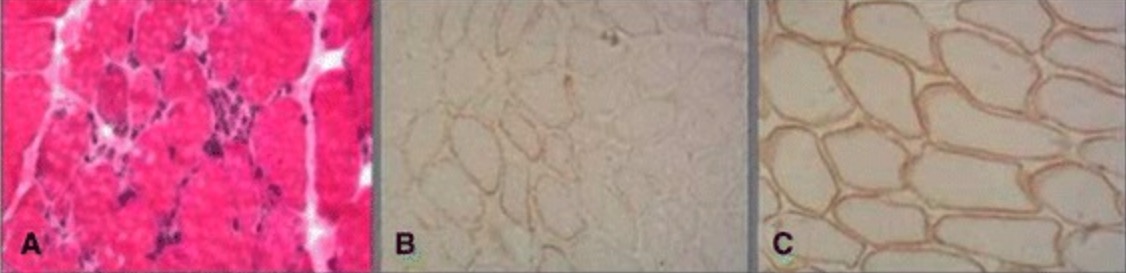
Muscle biopsy in Becker muscular dystrophy:
Histopathological examination shows necrotic muscle fibers with replacement by fibrous and adipose tissue (A). Immunostaining shows partial positivity for dystrophin in Becker muscular dystrohy (B) compared with normal muscle (C).
No curative treatment exists for BMD. Management is aimed at supportive and palliative care.