La distrofia muscular de Becker es un trastorno genético recesivo ligado al AL Amyloidosis cromosoma X que está causado por una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy. Se produce la proteina anormal y parcialmente funcional llamada distrofina, lo que lleva a la debilidad muscular progresiva y la eventual pérdida de la deambulación. El curso clínico es muy variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables, pero los LOS Neisseria síntomas suelen aparecer en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adolescencia. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las enzimas musculares, las pruebas genéticas y la biopsia muscular ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum caso de requerirse). El tratamiento es de soporte y tiene como objetivo frenar la progresión de la enfermedad y las complicaciones. La miocardiopatía dilatada es la principal causa de muerte.
Last updated: Dec 15, 2025
La distrofia muscular de Becker es la segunda forma más común de distrofia muscular.
La distrofia muscular de Becker está causada por mutaciones de duplicación o deleción sin desplazamiento del marco del gen DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el brazo corto del cromosoma X.
Fisiología normal:
Fisiopatología en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la Distrofia muscular de Becker:
La distrofia muscular de Becker tiene una presentación muy variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables.
La distrofia muscular de Becker sigue un curso de debilidad muscular lentamente progresiva en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un patrón de proximal a distal.
Síntomas:
Hallazgos al AL Amyloidosis examen físico:
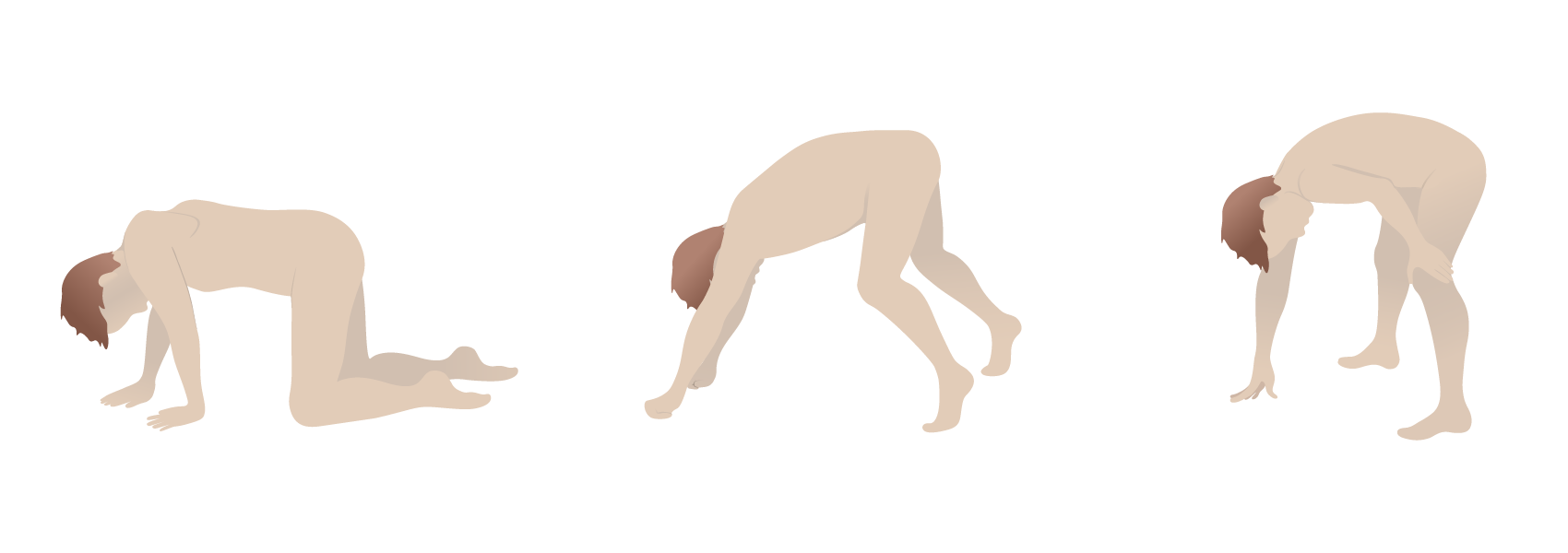
Signo de Gower: el uso de los brazos y las manos para maniobrar hasta una posición de pie debido a la debilidad muscular proximal
Imagen por Lecturio.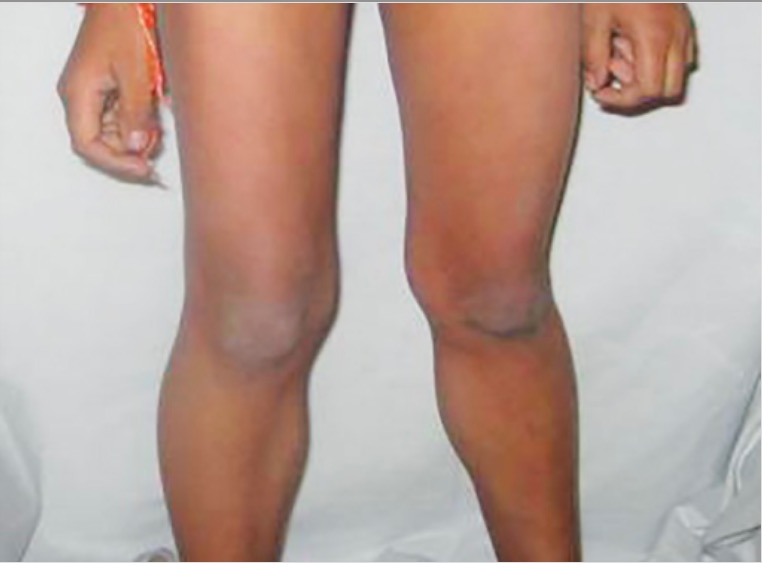
Pseudohipertrofia de la pantorrilla por distrofia muscular: músculos gastrocnemios grandes y débiles debido a la sustitución por grasa y tejido fibroso
Image: “Calf hypertrophy” por Professor, Department of Pedodontics and Preventive Dentistry, BJS Dental College, Ludhiana, Punjab, India. Licencia: CC BY 3.0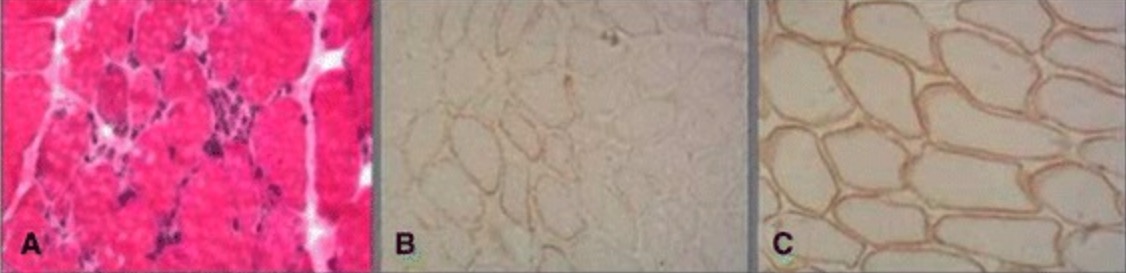
Biopsia muscular en la distrofia muscular de Becker:
El examen histopatológico muestra fibras musculares necróticas con sustitución por tejido fibroso y adiposo (A). La inmunotinción muestra positividad parcial para distrofina en distrofia muscular Becker (B) en comparación con el músculo normal (C).
No existe ningún tratamiento curativo para la distrofia muscular de Becker. El tratamiento está dirigido a los LOS Neisseria cuidados de apoyo y paliativos.