


A distrofia muscular de Becker (BMD) é uma doença genética recessiva ligado ao cromossoma X, causado por uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy. É produzida uma proteína da distrofina muscular anormal, parcialmente funcional, que leva à fraqueza muscular progressiva e à perda eventual de capacidade da marcha. A evolução clínica é altamente variável, mas os sintomas geralmente aparecem na adolescência. O diagnóstico é estabelecido com base nos valores de enzimas musculares, testes Testes Gonadal Hormones genéticos e biópsia muscular (se necessária). O tratamento da BMD é de suporte e visa retardar a progressão da doença assim como das suas complicações. A miocardiopatia dilatada é a principal causa de morte.
Last updated: Dec 15, 2025
A distrofia muscular de Becker (BMD) é a 2ª forma mais MAIS Androgen Insensitivity Syndrome comum de distrofia muscular.
A distrofia muscular de Becker é causada por uma mutação de deleção ou duplicação non-frameshift do gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy no braço curto do cromossoma X.
Fisiologia normal:
Fisiopatologia na BMD:
A distrofia muscular de Becker tem uma apresentação altamente variável.
A distrofia muscular de Becker cursa com fraqueza muscular lentamente progressiva, num padrão de proximal para distal.
Sintomas:
Achados do exame objetivo:
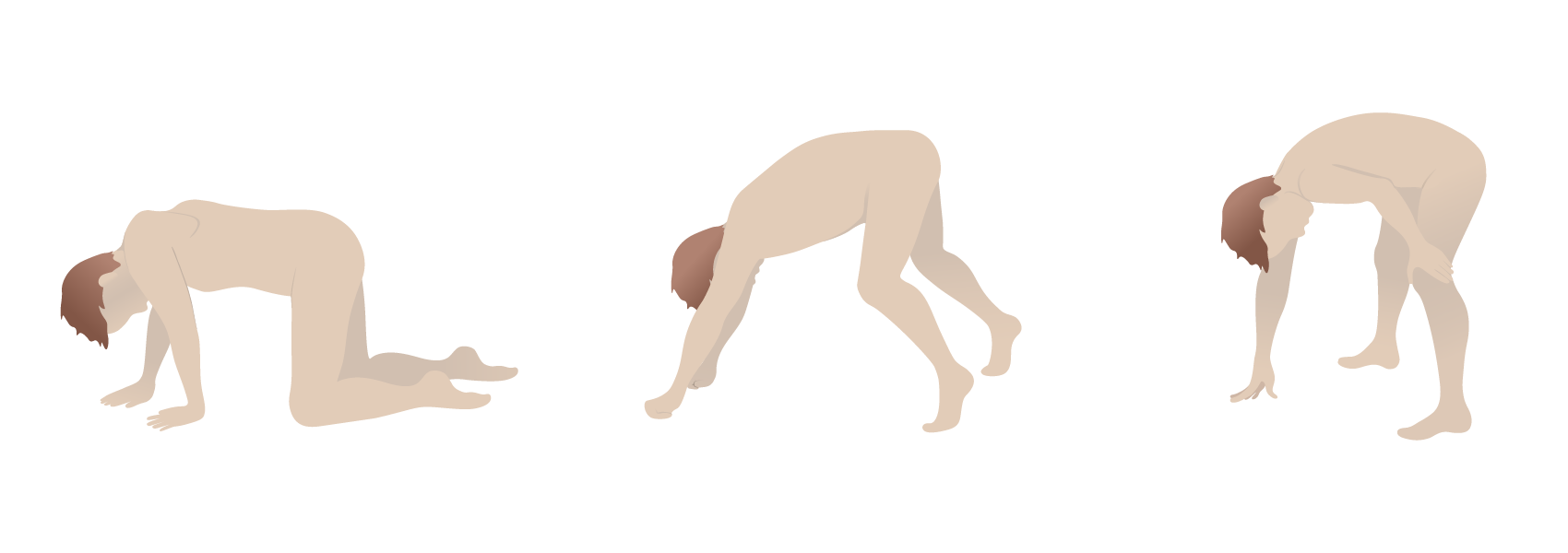
Sinal de Gower: devido à fraqueza muscular proximal, recorre-se ao uso dos braços e mãos como apoio para posicionamento em pé
Imagem por Lecturio.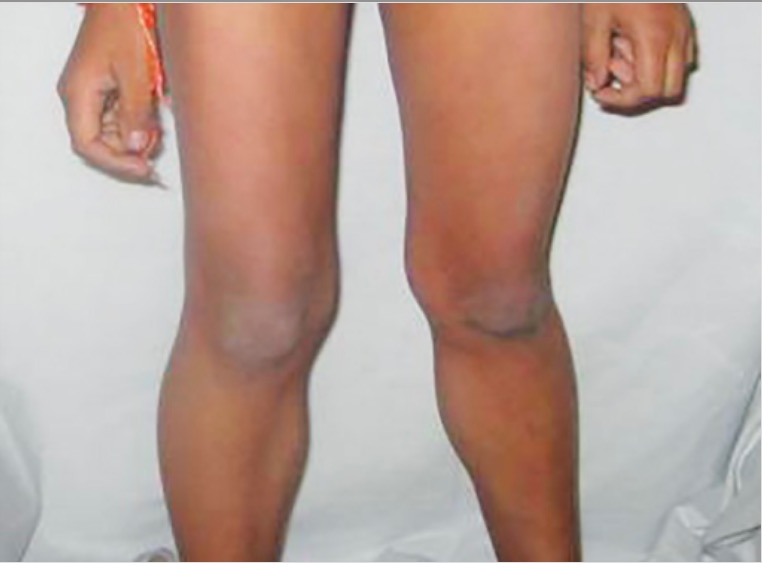
Pseudopertrofia dos músculos gastrocnémios devido à distrofia muscular: músculos grandes e fracos devido à substituição por gordura e tecido fibroso
Imagem: “Calf hypertrophy” por Professor, Departamento de Pedodontia e Odontologia Preventiva, Faculdade Dentária BJS, Ludhiana, Punjab, India. Licença: CC BY 3.0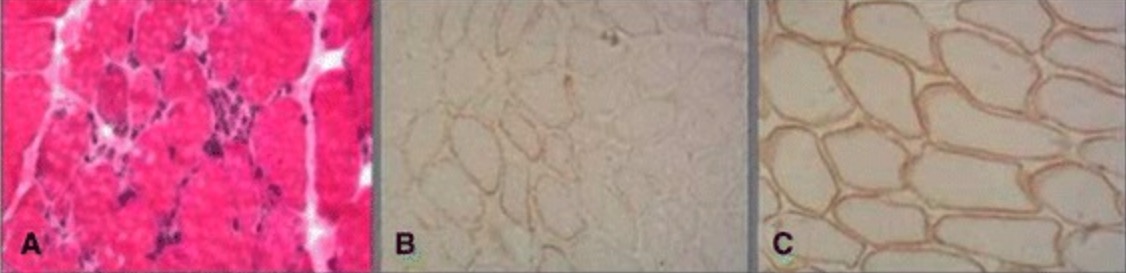
Biópsia muscular na distrofia muscular de Becker:
O exame histopatológico mostra fibras musculares necróticas com substituição por tecido fibroso e adiposo (A). A técnica de immunostaining mostra positividade parcial para a distrofina na distrofia muscular de Becker (B) em comparação com o músculo normal (C).
Não existe tratamento curativo para a BMD. O tratamento é dirigido a medidas de suporte e cuidados paliativos.
