Duchenne muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy (DMD) is an X-linked X-linked Genetic diseases that are linked to gene mutations on the X chromosome in humans or the X chromosome in other species. Included here are animal models of human X-linked diseases. Common Variable Immunodeficiency (CVID) recessive genetic disorder that is caused by a mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the DMD gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics. The mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations leads to the production of abnormal dystrophin Dystrophin A muscle protein localized in surface membranes which is the product of the Duchenne/Becker muscular dystrophy gene. Individuals with Duchenne muscular dystrophy usually lack dystrophin completely while those with Becker muscular dystrophy have dystrophin of an altered size. It shares features with other cytoskeletal proteins such as spectrin and alpha-actinin but the precise function of dystrophin is not clear. One possible role might be to preserve the integrity and alignment of the plasma membrane to the myofibrils during muscle contraction and relaxation. Blotting Techniques, resulting in muscle-fiber destruction and replacement with fatty or fibrous Fibrous Fibrocystic Change tissue. Affected individuals present with progressive proximal muscle weakness Proximal Muscle Weakness Lambert-Eaton Myasthenic Syndrome leading to the eventual loss of ambulation, as well as contractures Contractures Prolonged shortening of the muscle or other soft tissue around a joint, preventing movement of the joint. Wound Healing, scoliosis Scoliosis Scoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis, cardiomyopathy Cardiomyopathy Cardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types, and respiratory failure Respiratory failure Respiratory failure is a syndrome that develops when the respiratory system is unable to maintain oxygenation and/or ventilation. Respiratory failure may be acute or chronic and is classified as hypoxemic, hypercapnic, or a combination of the two. Respiratory Failure. A marked elevation in CK may be observed. Genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies is used to confirm the diagnosis. Management is supportive and aimed at slowing disease progression and complications. Duchenne muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy is fatal with a life expectancy Life expectancy Based on known statistical data, the number of years which any person of a given age may reasonably expected to live. Population Pyramids of about 20 years of age.
Last updated: May 16, 2024
Duchenne muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy (DMD) is the most common and most severe muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy.
Duchenne muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy results from a mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the dystrophin Dystrophin A muscle protein localized in surface membranes which is the product of the Duchenne/Becker muscular dystrophy gene. Individuals with Duchenne muscular dystrophy usually lack dystrophin completely while those with Becker muscular dystrophy have dystrophin of an altered size. It shares features with other cytoskeletal proteins such as spectrin and alpha-actinin but the precise function of dystrophin is not clear. One possible role might be to preserve the integrity and alignment of the plasma membrane to the myofibrils during muscle contraction and relaxation. Blotting Techniques (DMD) gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics on the short arm Arm The arm, or “upper arm” in common usage, is the region of the upper limb that extends from the shoulder to the elbow joint and connects inferiorly to the forearm through the cubital fossa. It is divided into 2 fascial compartments (anterior and posterior). Arm: Anatomy of the X chromosome X chromosome The female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species. Basic Terms of Genetics.
Normal physiology:
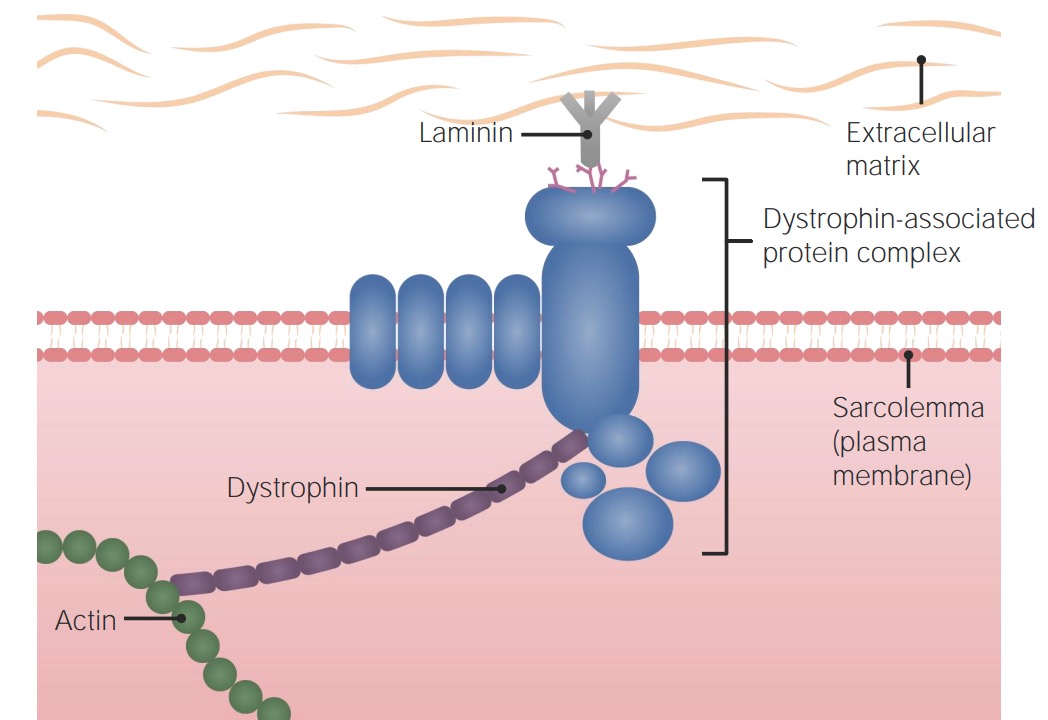
Normal muscle physiology:
Dystrophin protein helps link the dystrophin-associated protein complex (which links with the extracellular matrix) to the cytoskeleton of the muscle cell.
Pathophysiology of DMD:
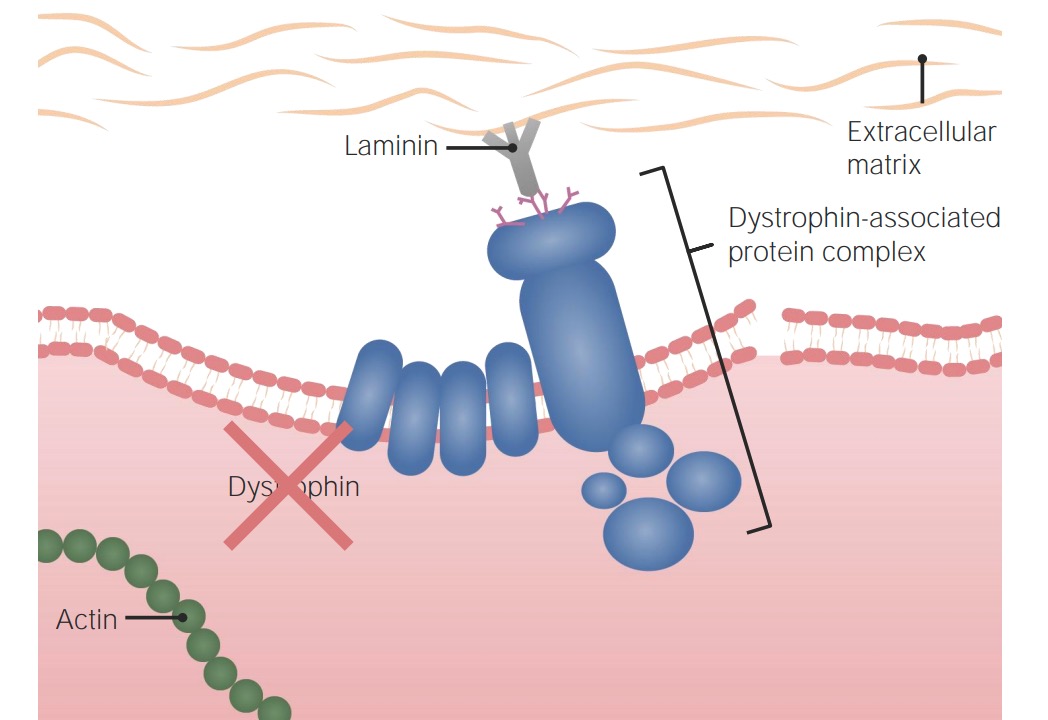
Pathogenesis of Duchenne muscular dystrophy:
Without a functional dystrophin protein, the cell is not appropriately anchored to the extracellular matrix and cellular cytoskeleton, rendering it susceptible to damage and death.
Duchenne muscular dystrophy Muscular Dystrophy Becker Muscular Dystrophy presents with progressive muscle weakness.
Timeline:
Clinical course:
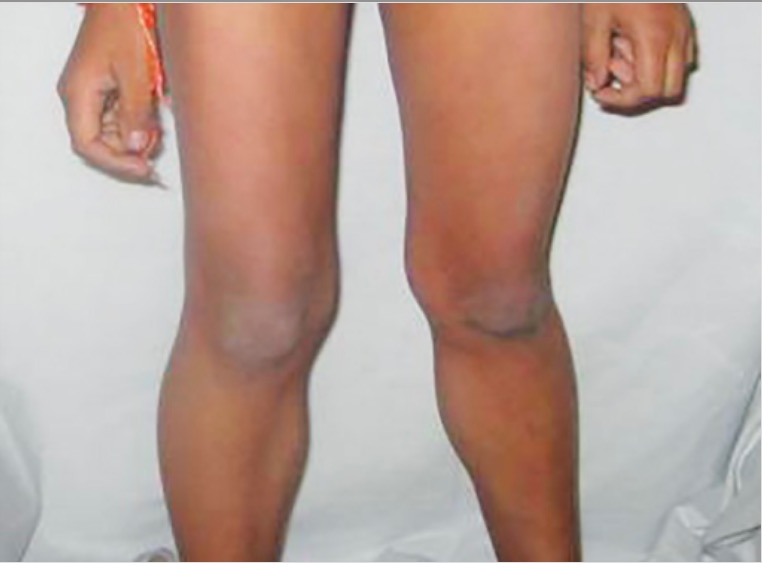
Calf pseudohypertrophy in Duchenne muscular dystrophy
Image: “Calf hypertrophy” by Professor, Department of Pedodontics and Preventive Dentistry, BJS Dental College, Ludhiana, Punjab, India. License: CC BY 3.0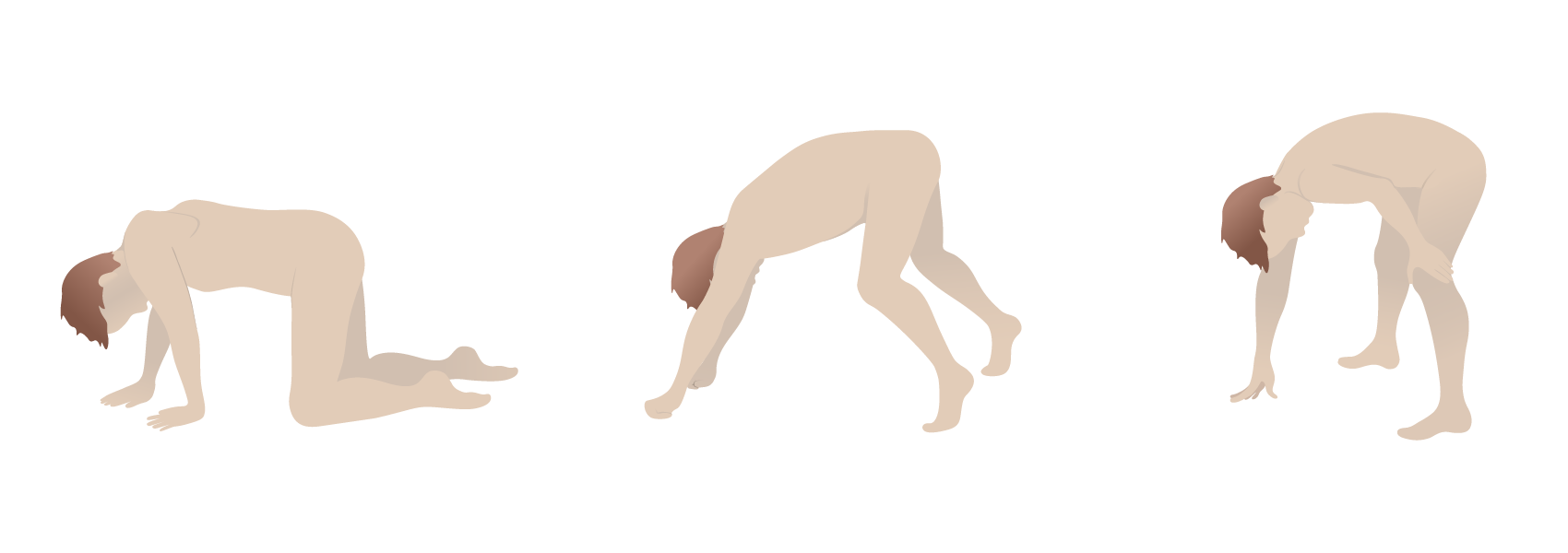
Gower’s sign in Duchenne muscular dystrophy: the use of the arms and hands to maneuver up to a standing position due to proximal muscle weakness
Image by Lecturio.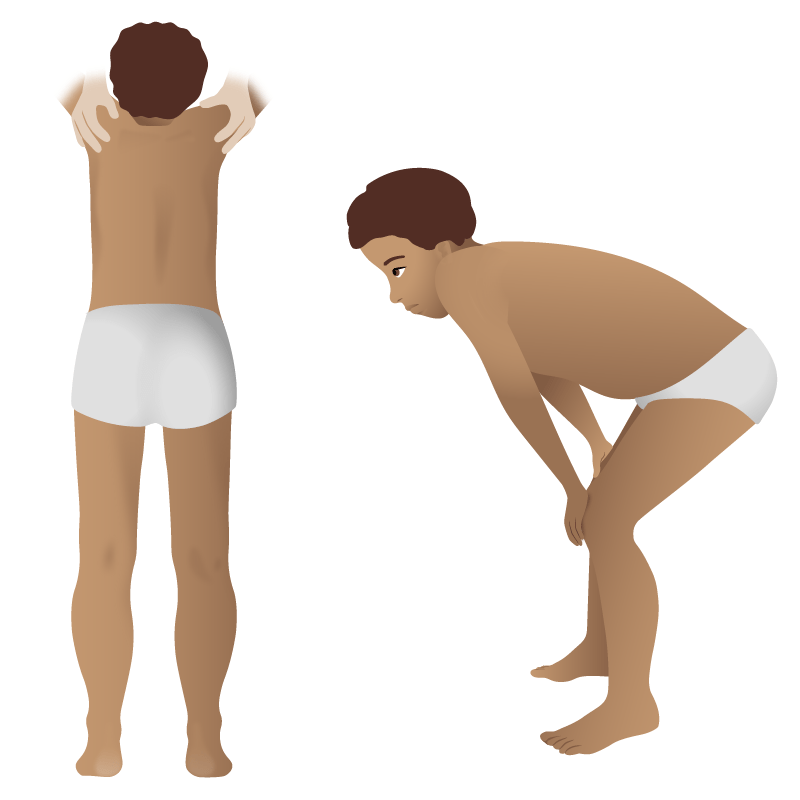
Physical findings in Duchenne muscular dystrophy:
These images are of a boy with proximal muscle weakness and calf pseudohypertrophy.
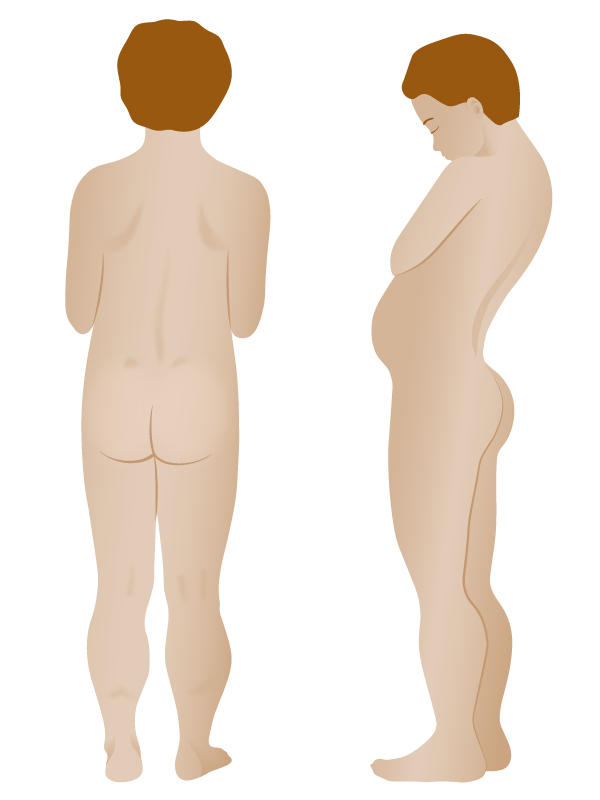
Drawing demonstrating physical findings in Duchenne muscular dystrophy:
There is excessive development of the calves (pseudohypertrophy) and thinness of the arms. In the figure on the right, lumbar lordosis is visible.
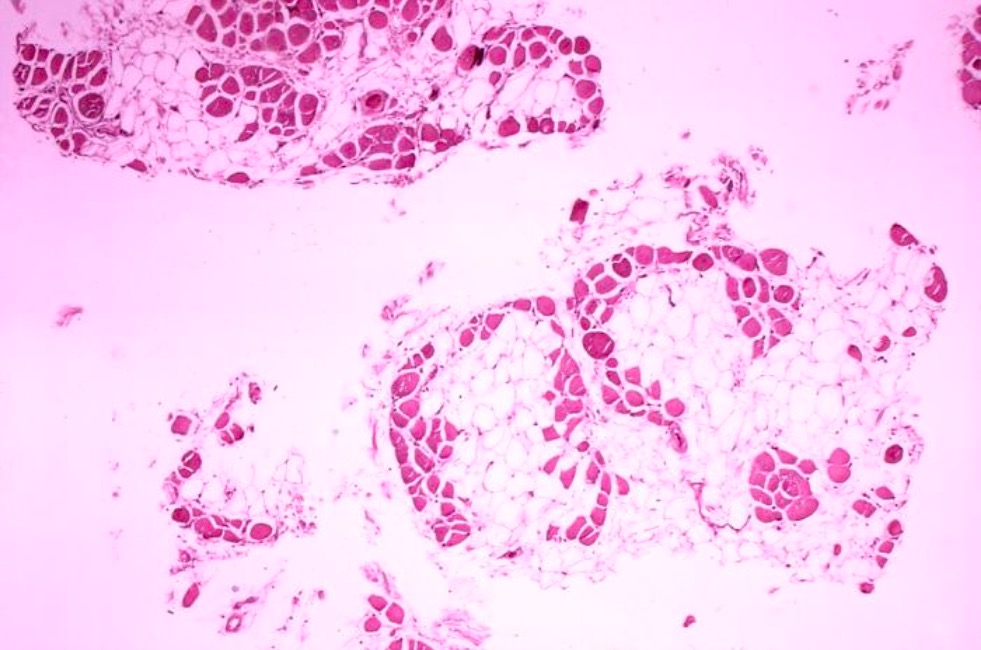
Histology image of muscle biopsy in Duchenne muscular dystrophy:
Significant replacement of muscle fibers with adipose cells (clear).
There is no cure for DMD. Management is mainly supportive and palliative, and often guided by individual preferences.