


A distrofia muscular de Duchenne ( DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy) é uma doença genética recessiva ligada ao cromossoma X, causada por uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy. Esta mutação leva a uma produção anormal de distrofina, o que resulta na destruição e substituição das fibras musculares em tecido adiposo ou fibroso. Os indivíduos afetados apresentam-se com fraqueza muscular proximal progressiva, que pode levar à perda da marcha, assim como contraturas musculares, escoliose, miocardiopatia e insuficiência respiratória. Pode-se ainda registar uma elevação acentuada da creatina cinase (CK). O diagnóstico é confirmado com o recurso a testes Testes Gonadal Hormones genéticos. A abordagem terapêutica consiste em medidas de suporte que visam retardar a progressão da doença e das suas complicações. A Distrofia muscular de Duchenne é fatal, apresentando uma esperança média de vida de cerca de 20 anos.
Last updated: Jun 30, 2022
A Distrofia muscular de Duchenne ( DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy) é a distrofia muscular mais MAIS Androgen Insensitivity Syndrome comum e mais MAIS Androgen Insensitivity Syndrome grave.
A Distrofia muscular de Duchenne resulta de uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics da distrofina ( DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy) no braço curto do cromossoma X.
Fisiologia normal:
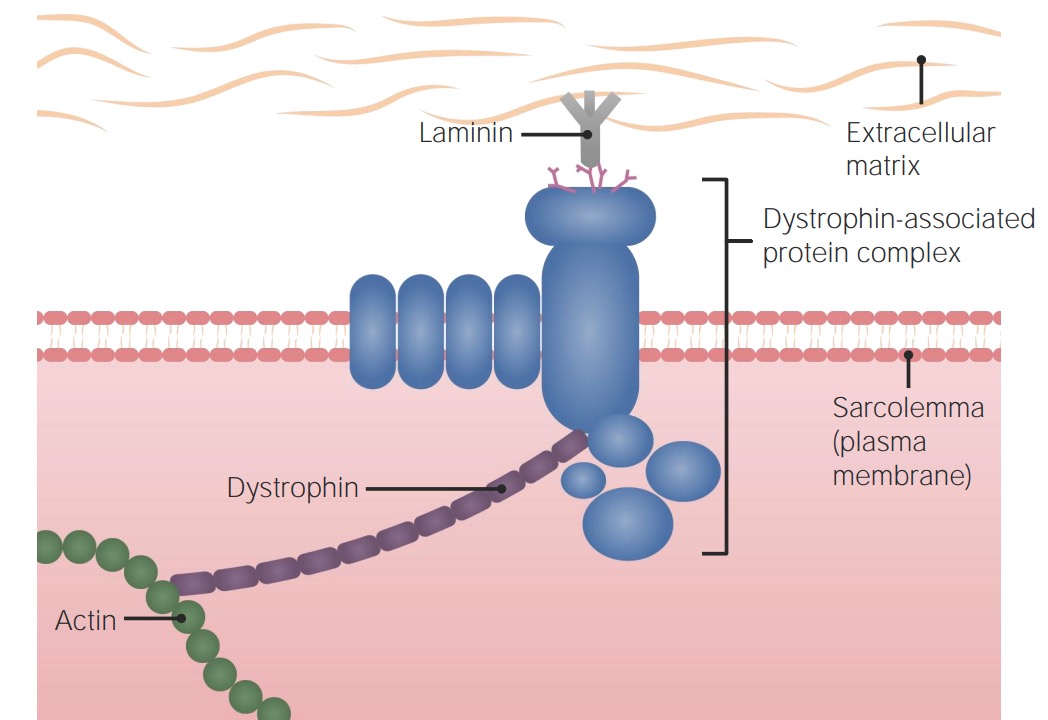
Fisiologia muscular normal:
A proteína da distrofina ajuda na ligação do complexo proteico associado à distrofina (que se conecta à matriz extracelular) ao citoesqueleto da célula muscular.
Fisiopatologia da DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy:
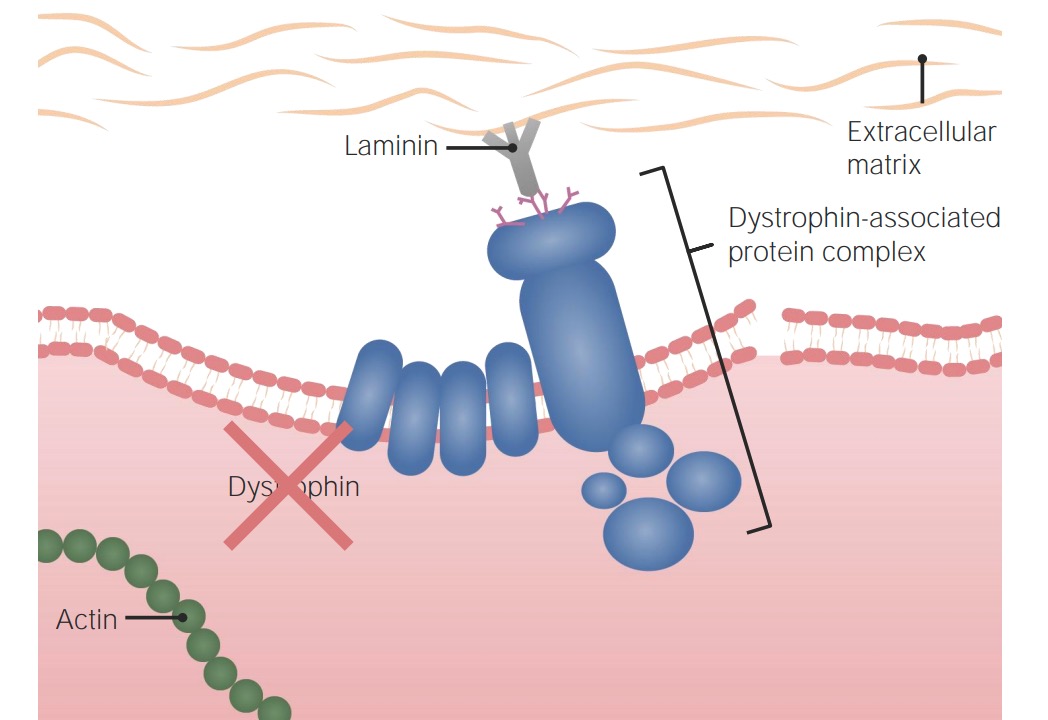
Patogénese da distrofia muscular de Duchenne:
Sem uma proteína da distrofina funcional, a célula não está adequadamente ancorada à matriz extracelular e ao citoesqueleto celular, tornando-a suscetível a dano e morte.
A distrofia muscular de Duchenne apresenta-se como fraqueza muscular progressiva.
Linha temporal:
Evolução clínica:
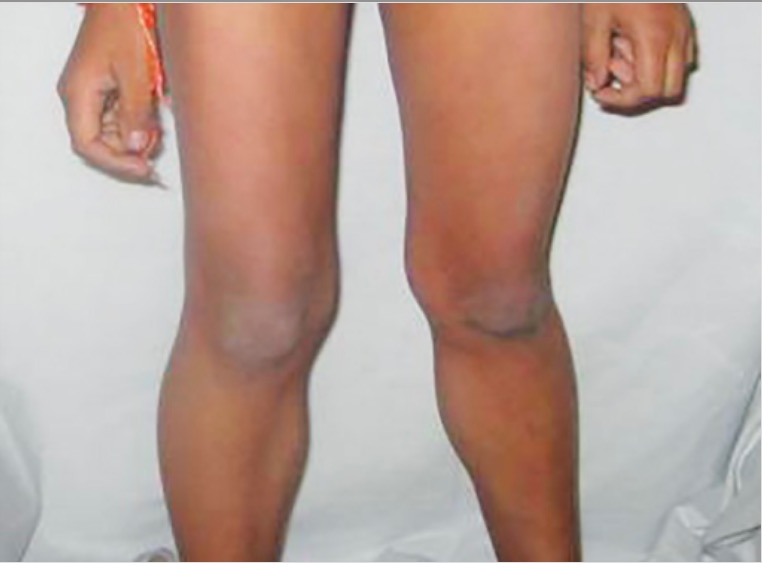
Pseudo-hipertrofia dos músculos gastrocnémios na distrofia muscular de Duchenne
Imagem: “Calf hypertrophy” por Professor, Departamento de Pedodontia e Odontologia Preventiva, Faculdade Dentária BJS, Ludhiana, Punjab, India. Licença: CC BY 3.0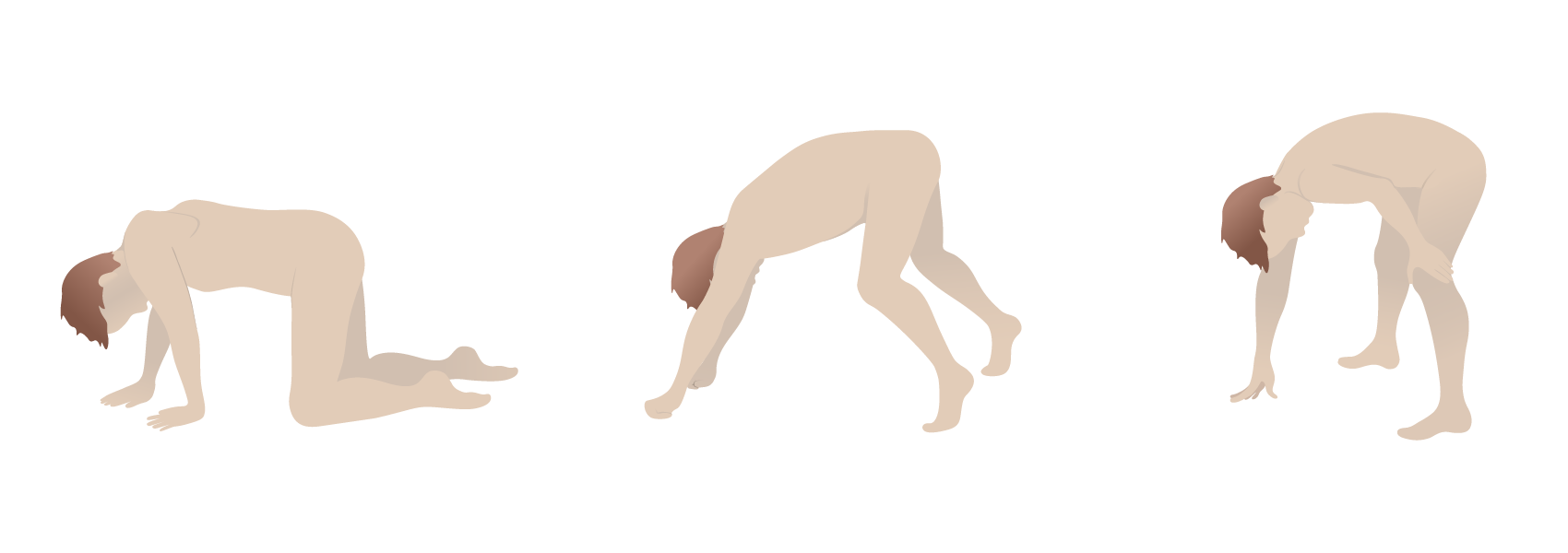
Sinal de Gower na distrofia muscular de Duchenne: devido à fraqueza muscular proximal, recorre-se ao uso dos braços e mãos como apoio para posicionamento em pé
Imagem por Lecturio.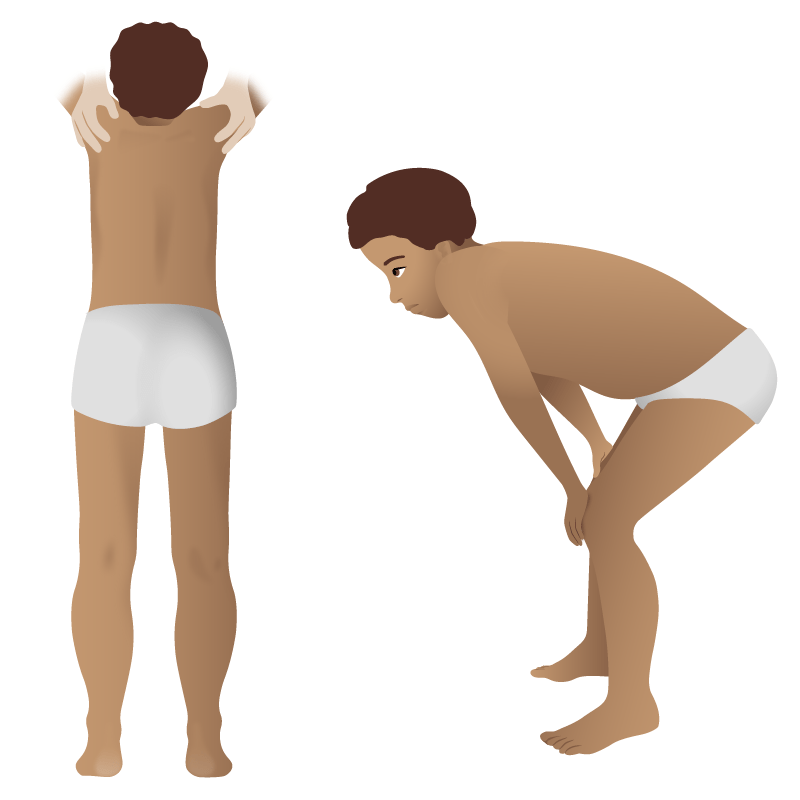
Achados ao exame objetivo na distrofia muscular de Duchenne:
Estas são imagens de um rapaz com fraqueza muscular proximal e pseudohipertrofia dos gastrocnémios.
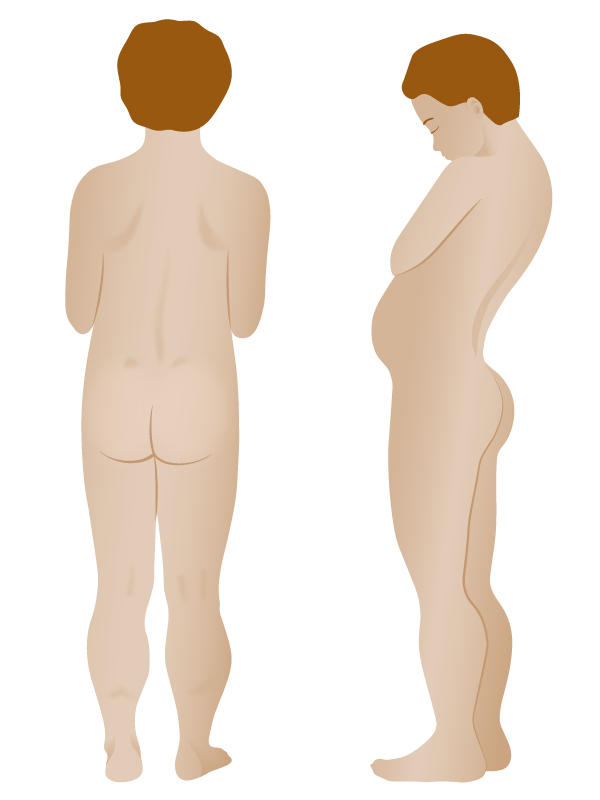
Desenho que mostra achados ao exame objetino na distrofia muscular de Duchenne:
Há desenvolvimento excessivo da região gemelar (pseudohipertrofia)e braços finos. Na figura da direita, a lordose lombar é visível.
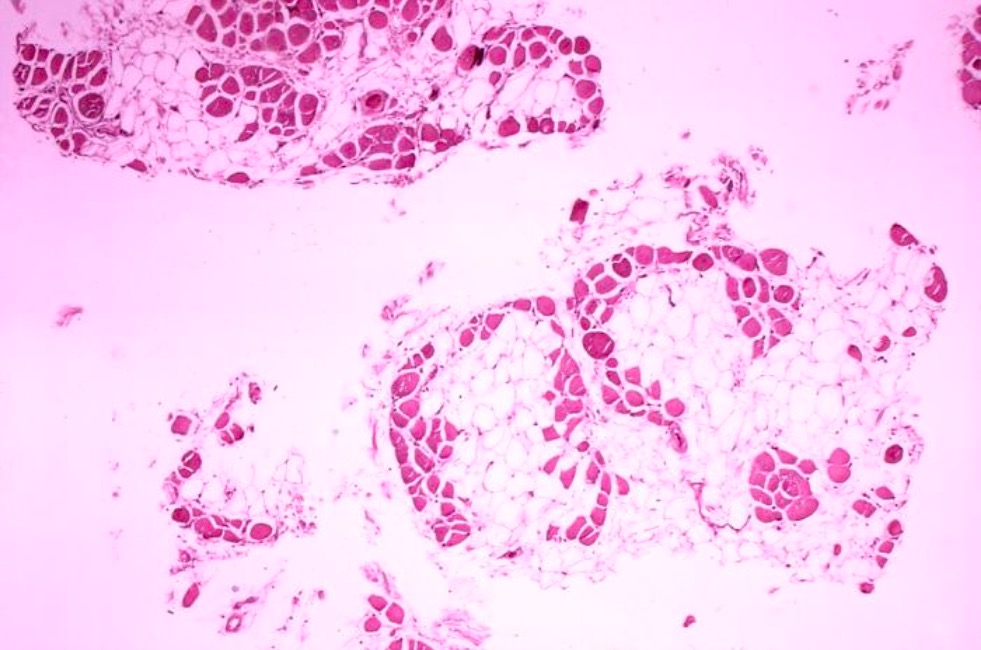
Imagem histológica da biópsia muscular na distrofia muscular de Duchenne:
Substituição significativa das fibras musculares por células adiposas (zonas a claro).
Não existe cura para a DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy. O tratamento é principalmente de suporte e paliativo, muitas vezes orientado pelas preferências individuais.
