La distrofia muscular de Duchenne es un trastorno genético recesivo ligado al AL Amyloidosis cromosoma X que está causado por una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy. La mutación lleva a la producción de distrofina anormal, dando como resultado la destrucción de la fibra muscular y el reemplazo con tejido graso o fibroso. Los LOS Neisseria individuos afectados presentan una debilidad muscular proximal progresiva que conduce a la pérdida de la deambulación, así como contracturas, escoliosis, cardiomiopatía e insuficiencia respiratoria. Puede observarse una marcada elevación de la CK. Las pruebas genéticas se utilizan para confirmar el diagnóstico. El tratamiento es de soporte y está dirigido a frenar la progresión de la enfermedad y las complicaciones. La distrofia muscular de Duchenne es terminal, con una esperanza de vida de unos 20 años.
Last updated: Jul 2, 2022
La distrofia muscular de Duchenne es la distrofia muscular más común y más grave.
La distrofia muscular de Duchenne es el resultado de una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen de la distrofina ( DMD DMD Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder that is caused by a mutation in the dmd gene. The mutation leads to the production of abnormal dystrophin, resulting in muscle-fiber destruction and replacement with fatty or fibrous tissue. Duchenne Muscular Dystrophy) en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el brazo corto del cromosoma X.
Fisiología normal:
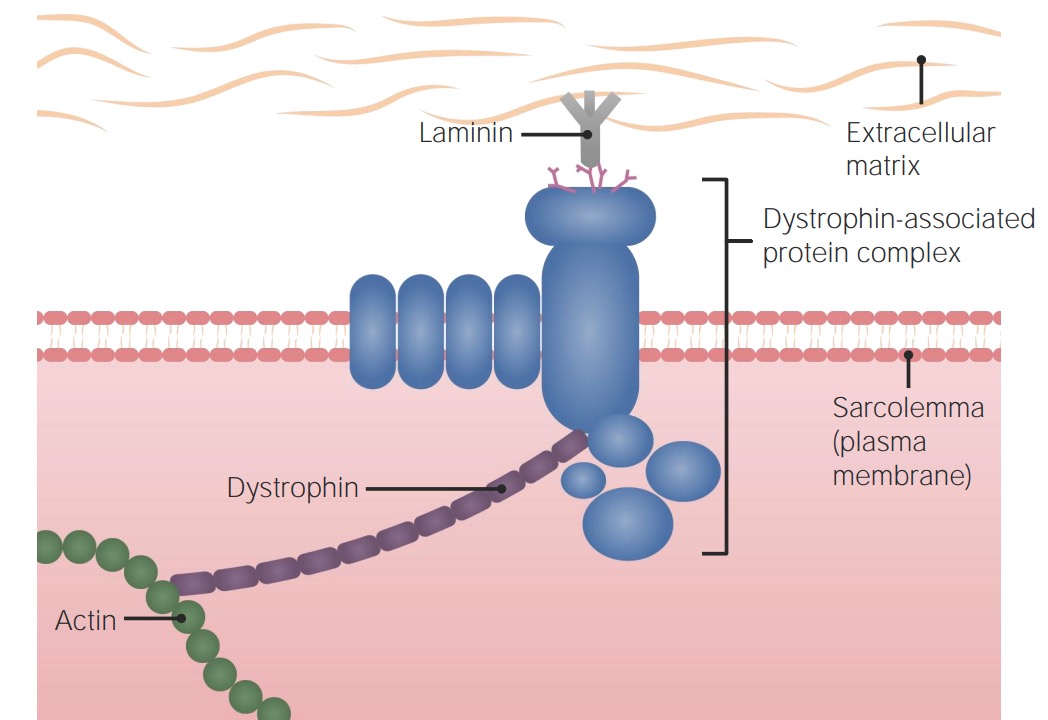
Fisiología muscular normal:
La proteína distrofina ayuda a enlazar el complejo de la proteína asociada a la distrofina (que se une con la matriz extracelular) al citoesqueleto de la célula muscular.
Fisiopatología de la distrofia muscular de Duchenne:
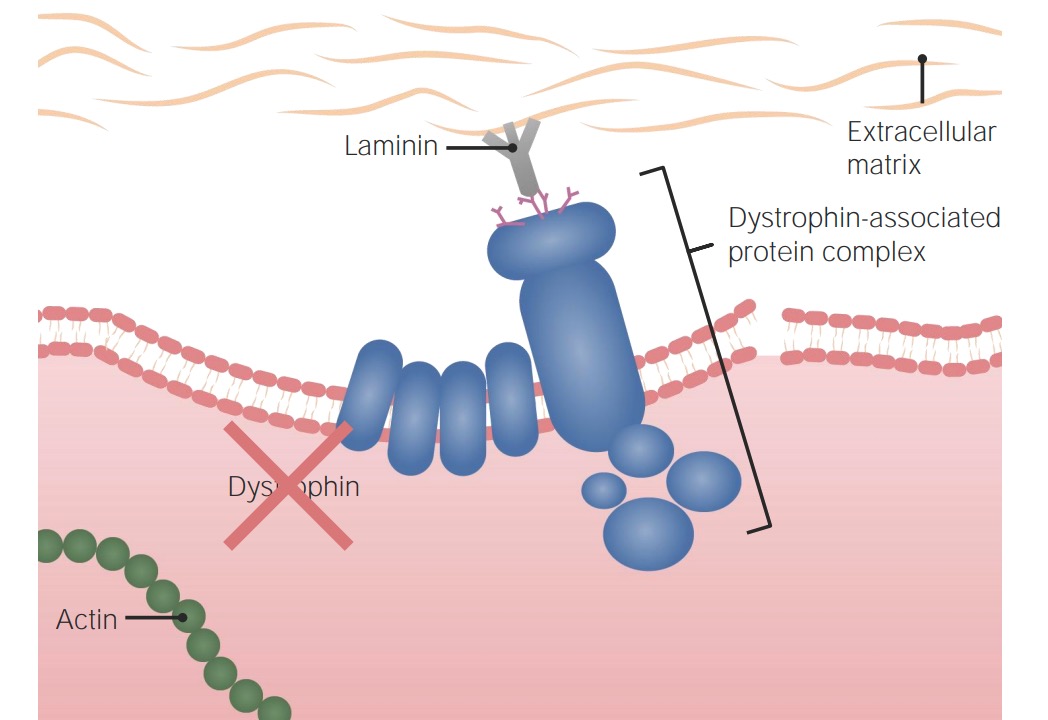
Patogénesis de la Distrofia muscular de Duchenne:
Sin una proteína distrofina funcional, la célula no está apropiadamente anclada a la matriz extracelular y al citoesqueleto celular, haciéndola susceptible al daño y la necrosis.
La distrofia muscular de Duchenne se presenta con una debilidad muscular progresiva.
Linea de tiempo:
Curso clínico:
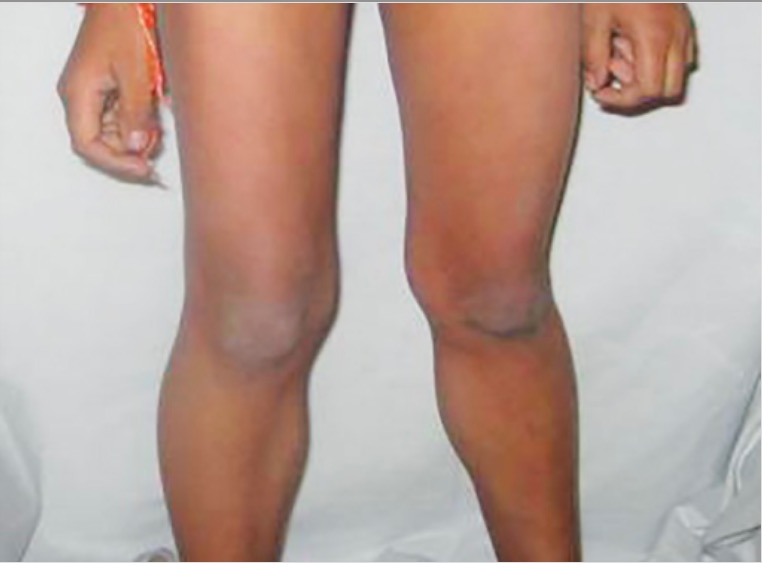
Pseudohipertrofia de pantorrilla en la distrofia muscular de Duchenne
Imagen: “Calf hypertrophy” por Professor, Department of Pedodontics and Preventive Dentistry, BJS Dental College, Ludhiana, Punjab, India. Licencia: CC BY 3.0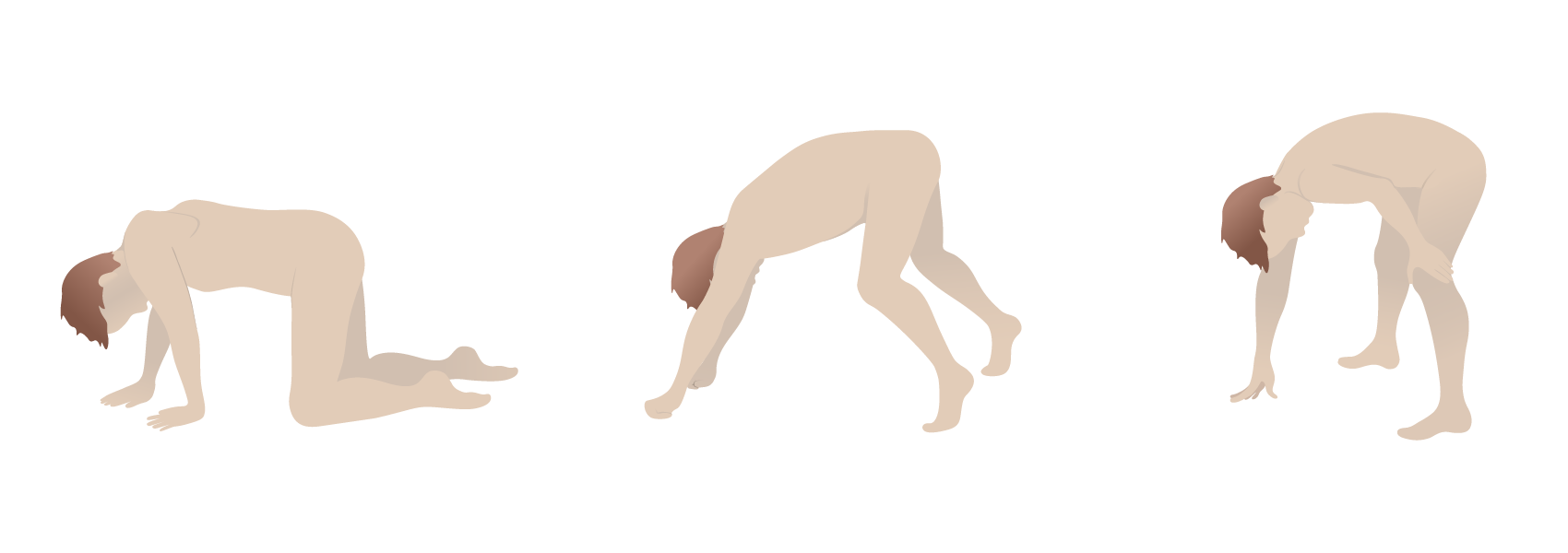
Signo de Gower en la distrofia muscular de Duchenne: el uso de los brazos y las manos para maniobrar hasta la posición de pie debido a la debilidad muscular proximal
Imagen por Lecturio.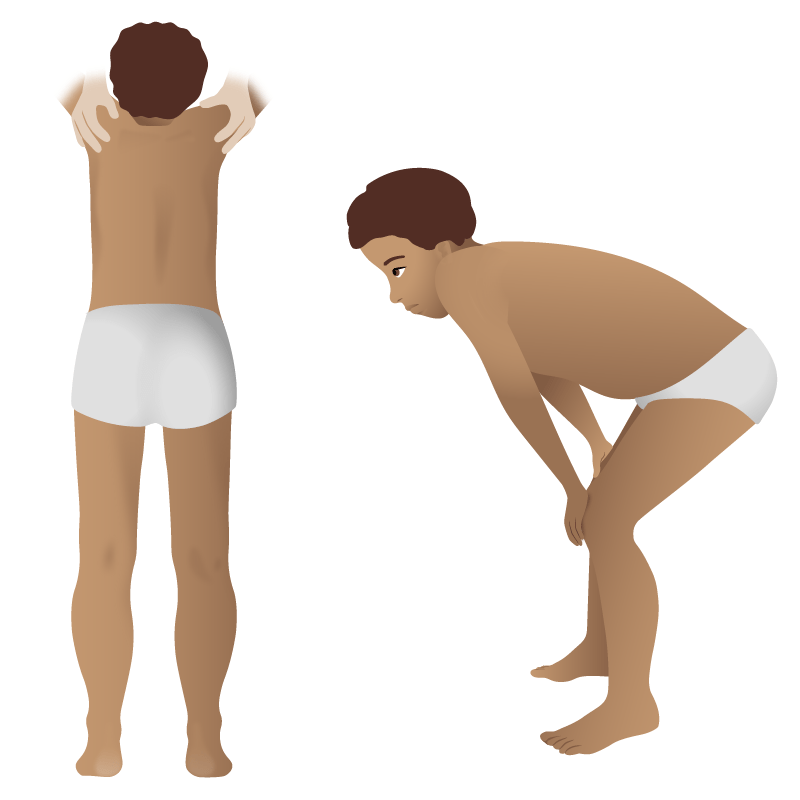
Hallazgos físicos en la distrofia muscular de Duchenne:
Estas imágenes son de un niño con debilidad muscular proximal y pseudohipertrofia de la pantorrilla.
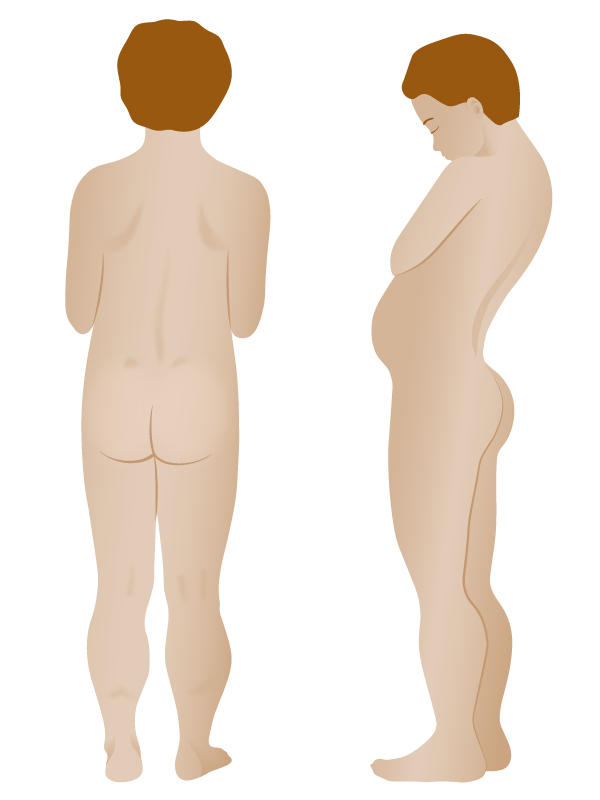
Dibujo que muestra los hallazgos físicos de la distrofia muscular de Duchenne:
Hay un desarrollo excesivo de las pantorrillas (pseudohipertrofia) y una delgadez de los brazos. En la figura de la derecha se aprecia lordosis lumbar.
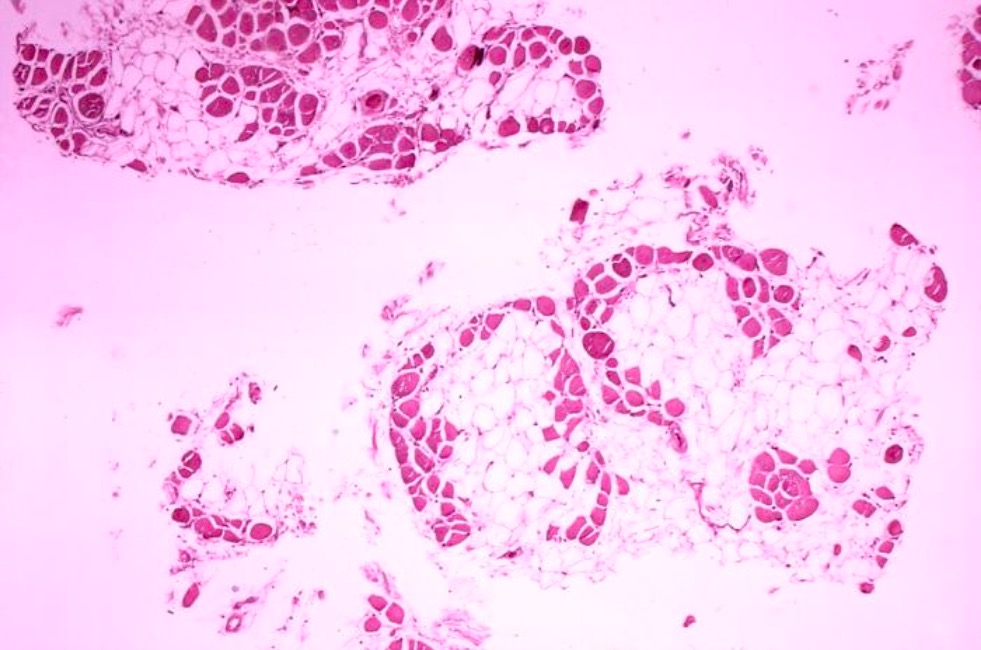
Imagen histológica de la biopsia muscular en la distrofia muscular de Duchenne:
Sustitución significativa de las fibras musculares por células adiposas (claro).
No hay cura para la distrofia muscular de Duchenne. El tratamiento es principalmente de soporte y paliativo, a menudo es guiado por preferencias individuales.