Proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis are 1 of the 3 primary macronutrients in the body and are synthesized from individual building blocks called amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids (AAs). Amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids are bound together by peptide bonds, which link the amino end of one AA AA Amyloidosis to the carboxy end of the next AA AA Amyloidosis, generating a protein's primary structure. The strand of AAs then undergoes additional folding, ultimately generating complex 3-dimensional structures. Proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis have a wide variety of functions, including catalytic, structural, regulatory, transport, storage, and immunologic functions. They are digested by proteases and peptidases secreted by the stomach Stomach The stomach is a muscular sac in the upper left portion of the abdomen that plays a critical role in digestion. The stomach develops from the foregut and connects the esophagus with the duodenum. Structurally, the stomach is C-shaped and forms a greater and lesser curvature and is divided grossly into regions: the cardia, fundus, body, and pylorus. Stomach: Anatomy and pancreas Pancreas The pancreas lies mostly posterior to the stomach and extends across the posterior abdominal wall from the duodenum on the right to the spleen on the left. This organ has both exocrine and endocrine tissue. Pancreas: Anatomy and absorbed as individual AAs in the small intestines through specialized transporters. There are countless medical conditions related to protein abnormalities, including abnormalities related to enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes, receptors Receptors Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors, membrane channels Channels The Cell: Cell Membrane, hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types, accumulation of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis, and autoimmune disorders.
Last updated: Dec 15, 2025
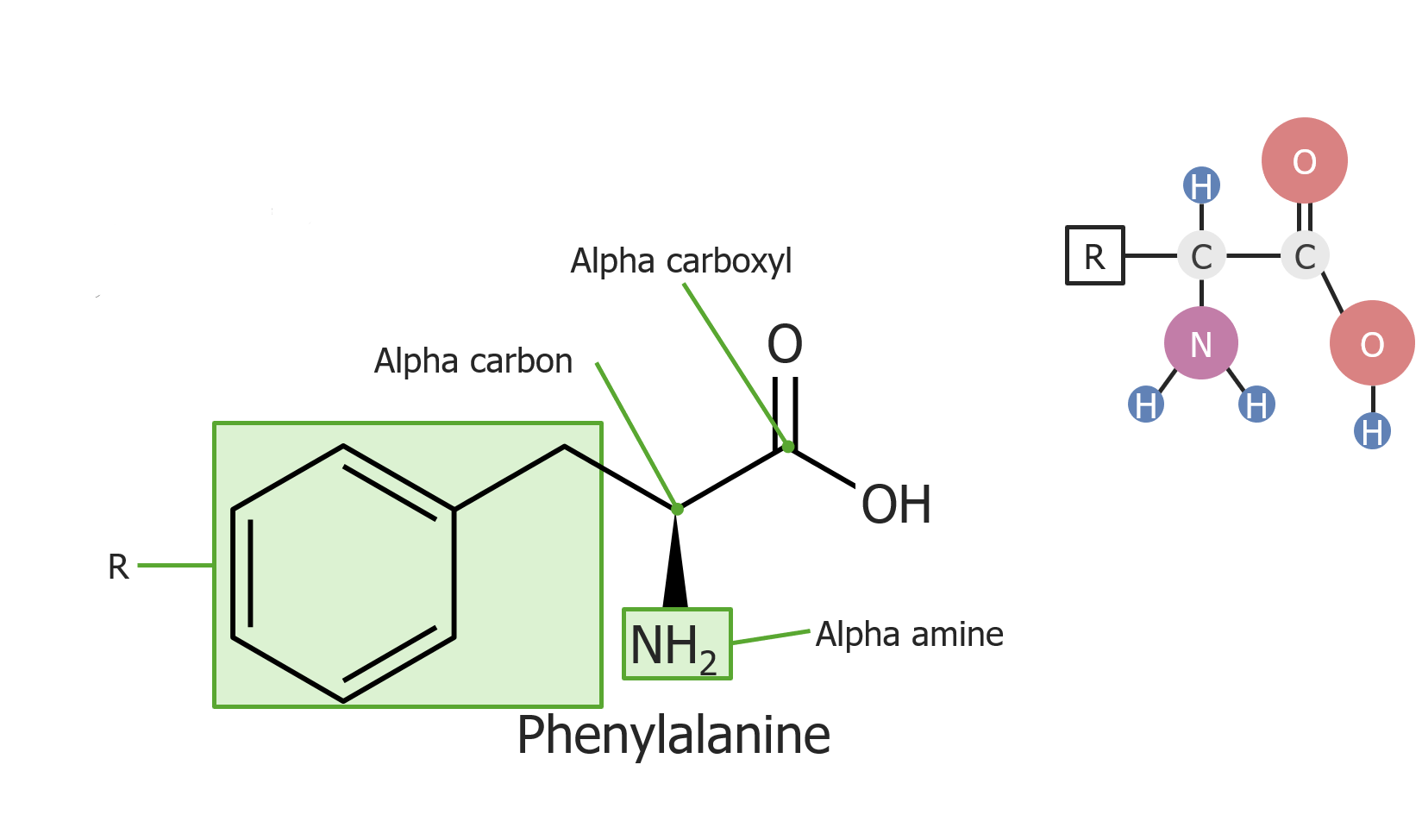
Example of the amino acid phenylalanine
Image by Lecturio.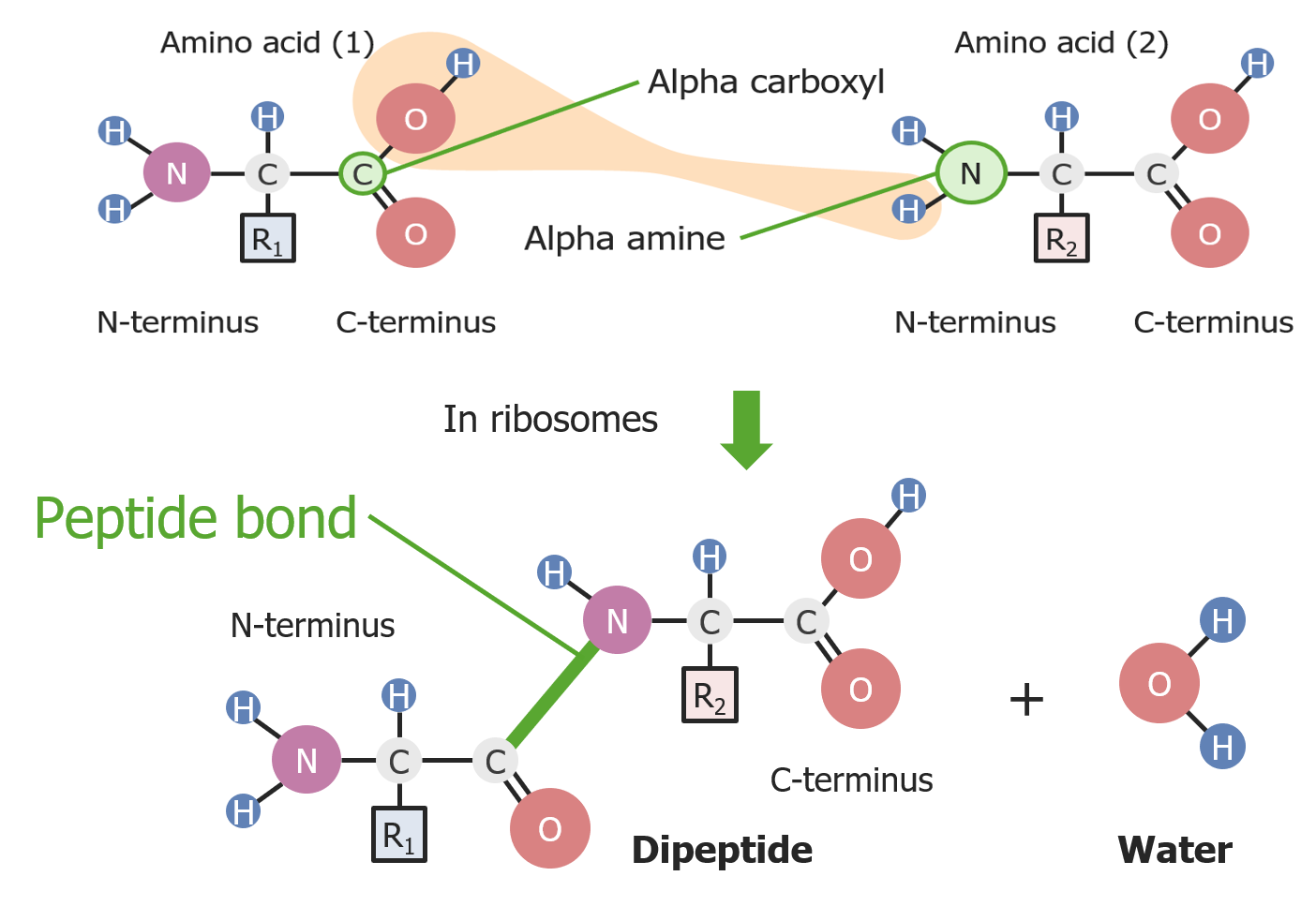
Formation of a peptide bond between 2 amino acids
Image by Lecturio.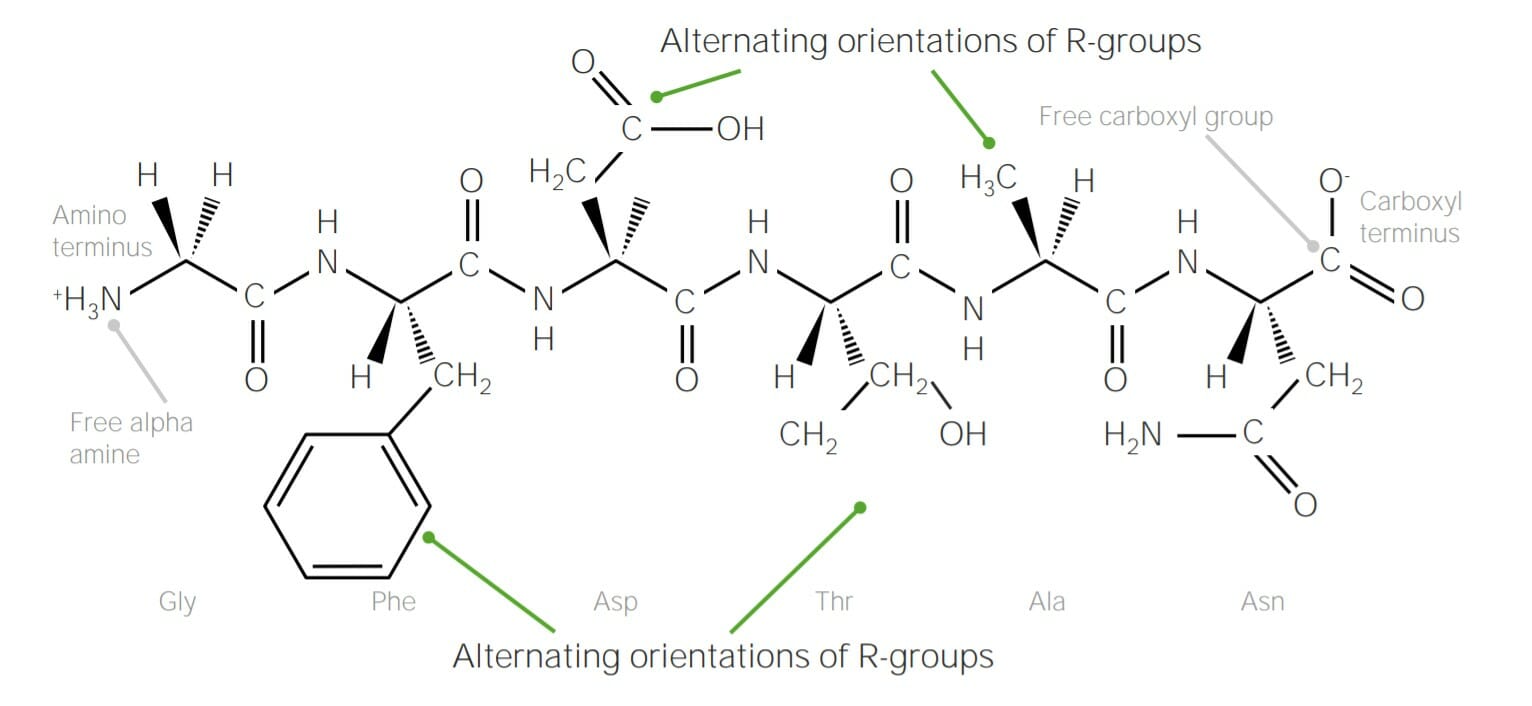
Example of polypeptide containing 6 amino acids, joined together with peptide bonds:
Note how the R groups of each amino acid alternate “sides” of the polypeptide chain; this is due to trans configuration of peptide bonds.
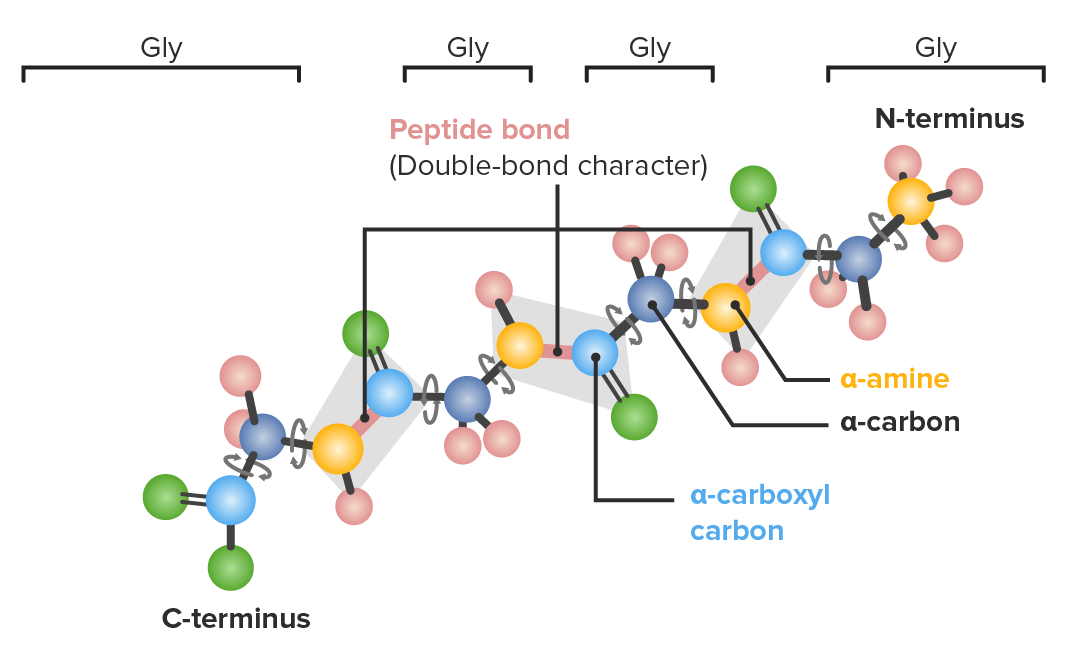
Example of a polypeptide with 4 glycine (gly) amino acids in sequence demonstrating which bonds have freedom to rotate:
Dark blue: α-carbons
Light blue: carboxyl carbons
Yellow: nitrogen
Green: oxygen
Pink: hydrogen
There are 4 levels of protein structure; this is often referred to as protein folding. The levels are primary, secondary, tertiary, and quaternary structures. Proper folding requires the assistance of chaperone proteins Chaperone proteins A family of cellular proteins that mediate the correct assembly or disassembly of polypeptides and their associated ligands. Although they take part in the assembly process, molecular chaperones are not components of the final structures. Post-translational Protein Processing
Primary structure:
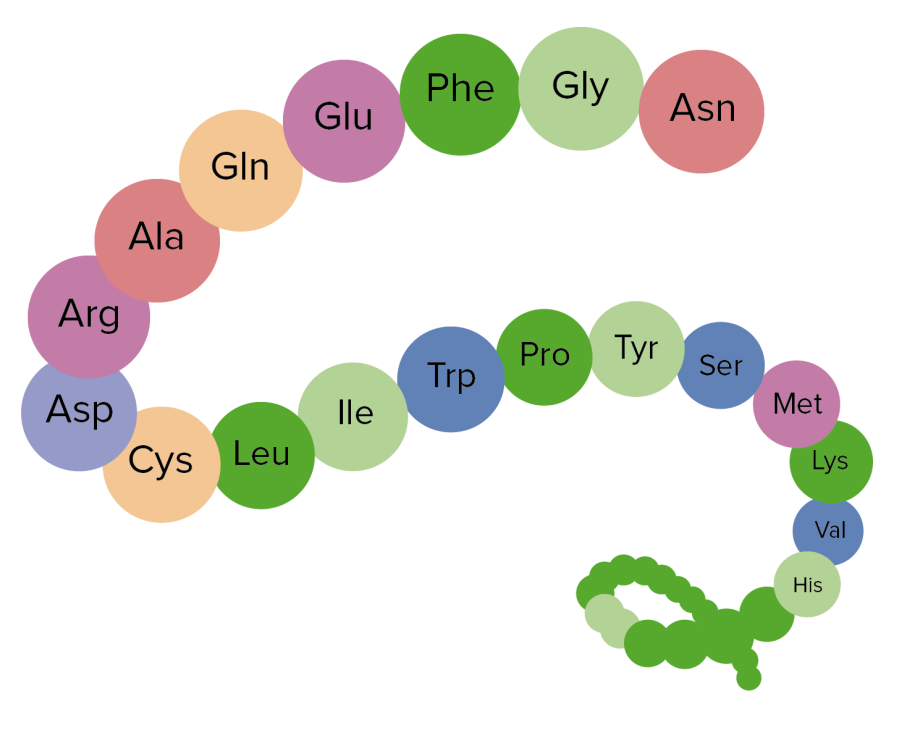
Image showing the primary structure of proteins, an aggregation of amino acids
Image by Lecturio.Secondary structure:
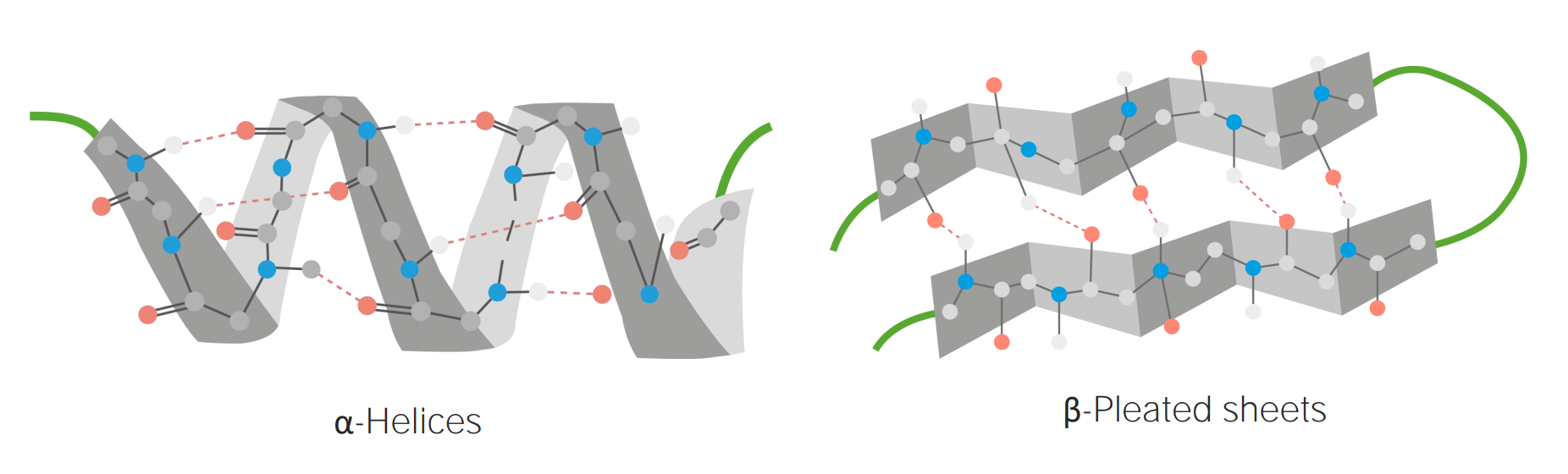
Examples of α-helices and β-pleated sheets
Image by Lecturio.Tertiary structure:

Example of tertiary structure
Image by Lecturio.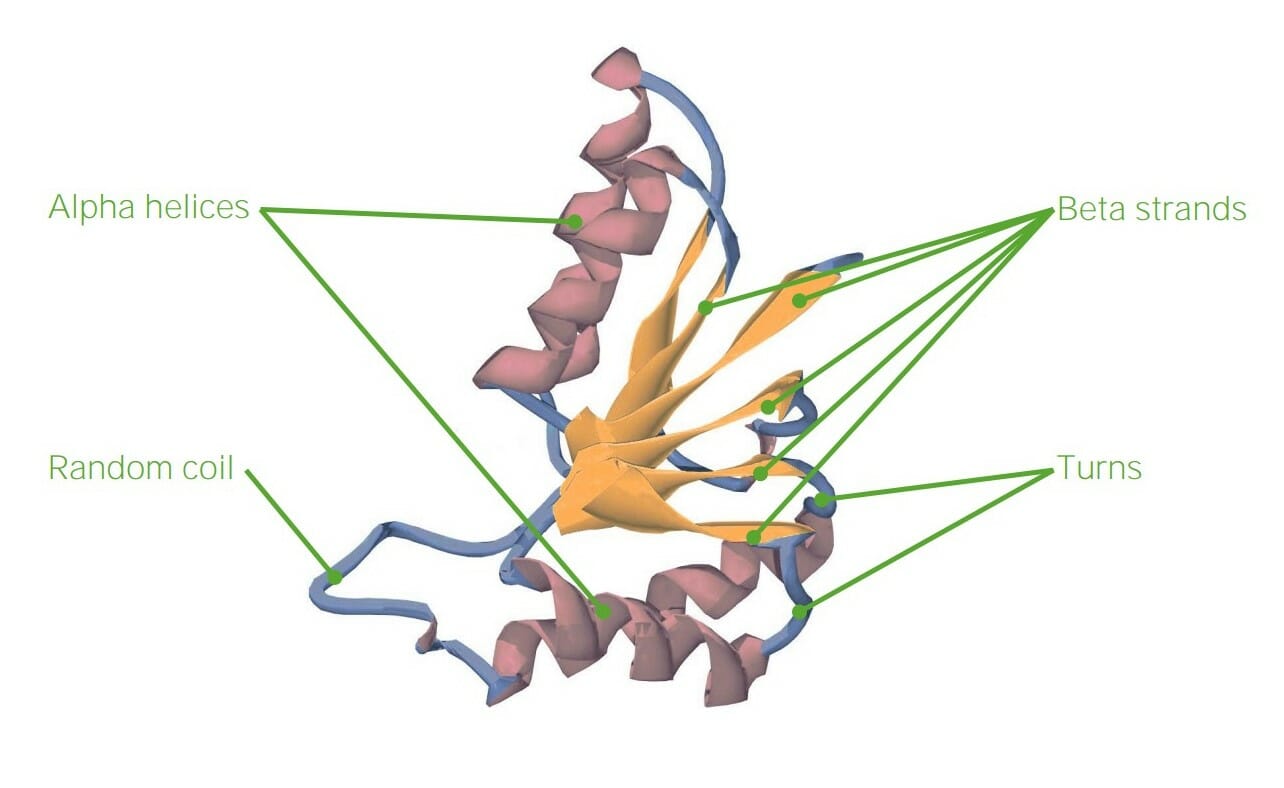
Image representing the tertiary structure of proteins
Image by Lecturio.Quaternary structure:
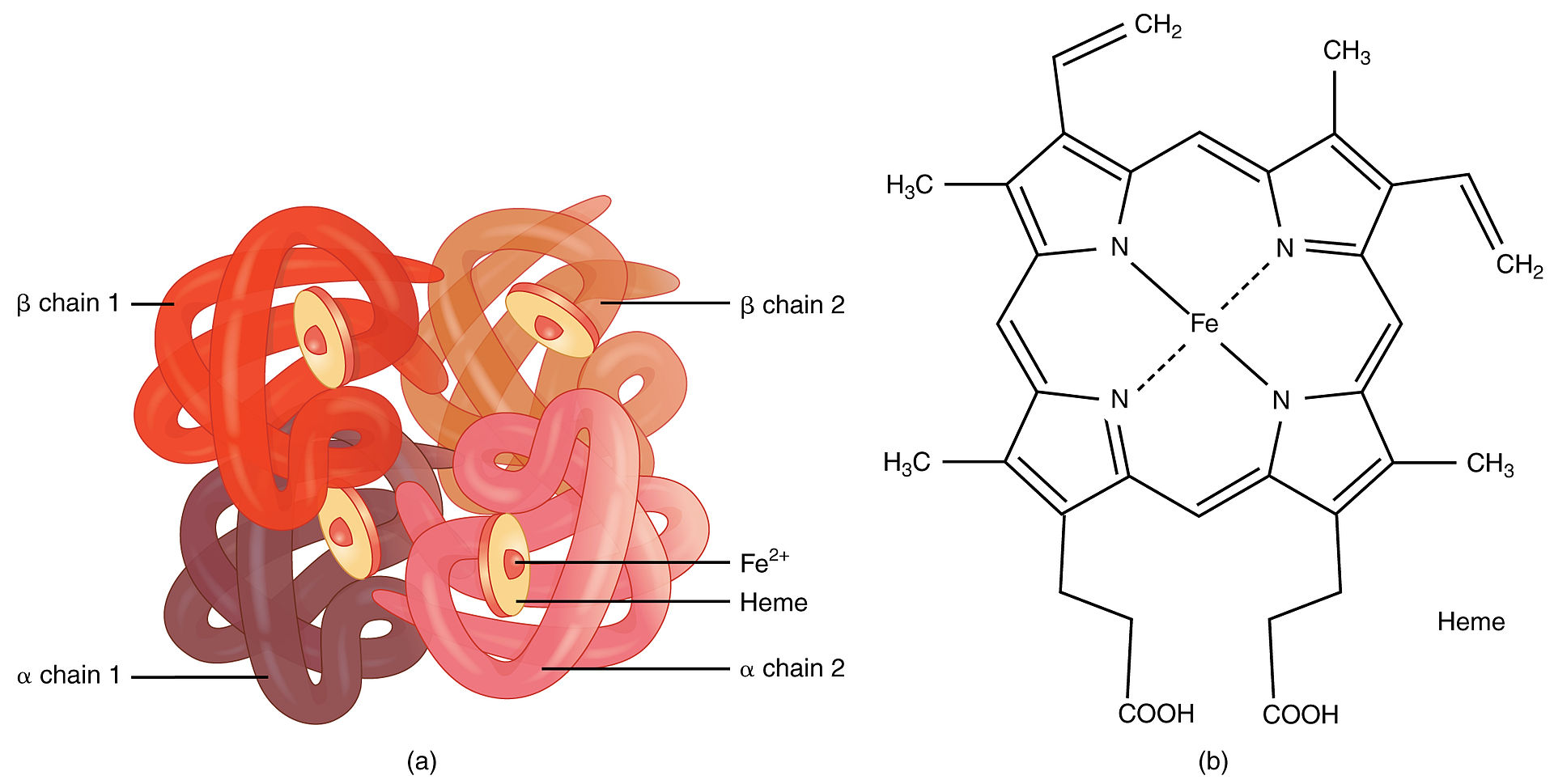
Hemoglobin:
An example of quaternary structure
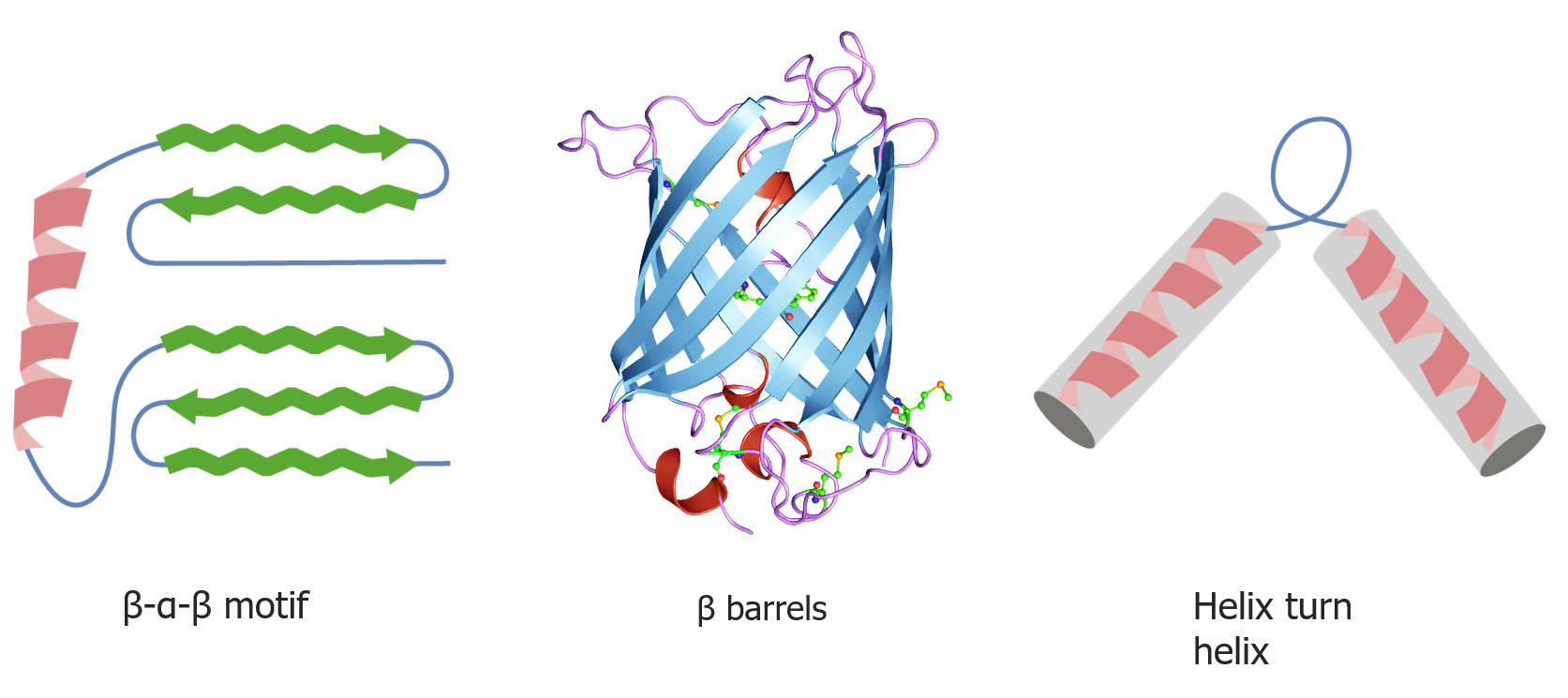
Quaternary and tertiary protein folding motifs
Image by Lecturio.Chaperone proteins Chaperone proteins A family of cellular proteins that mediate the correct assembly or disassembly of polypeptides and their associated ligands. Although they take part in the assembly process, molecular chaperones are not components of the final structures. Post-translational Protein Processing assist in protein folding.
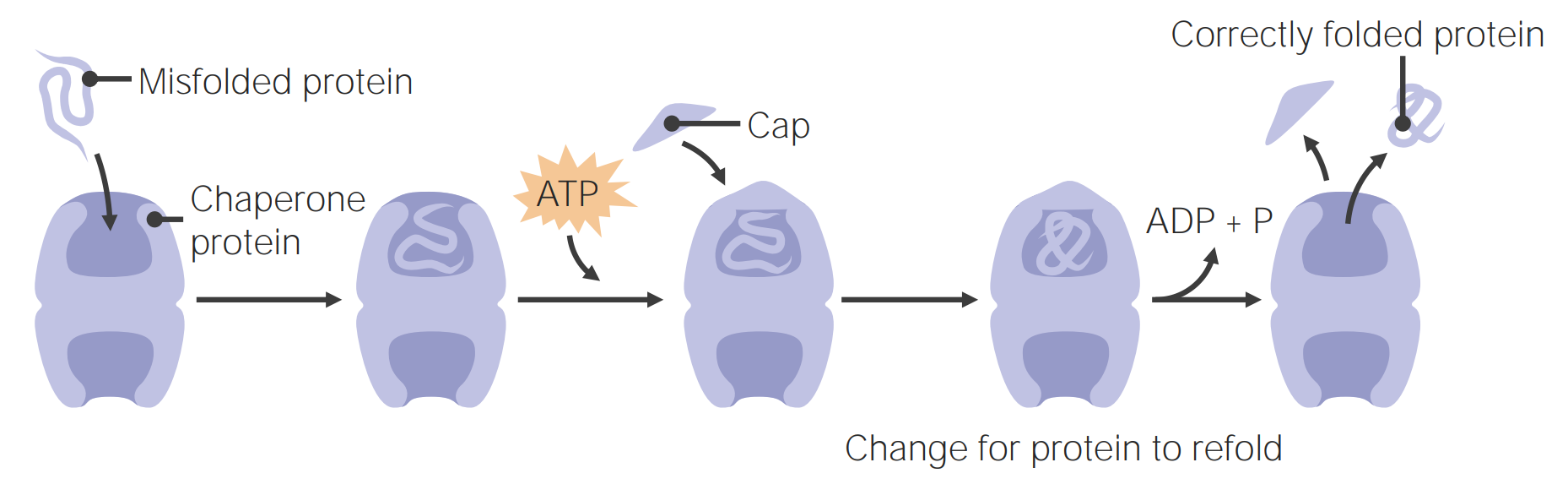
Chaperone proteins assist in protein folding
Image by Lecturio.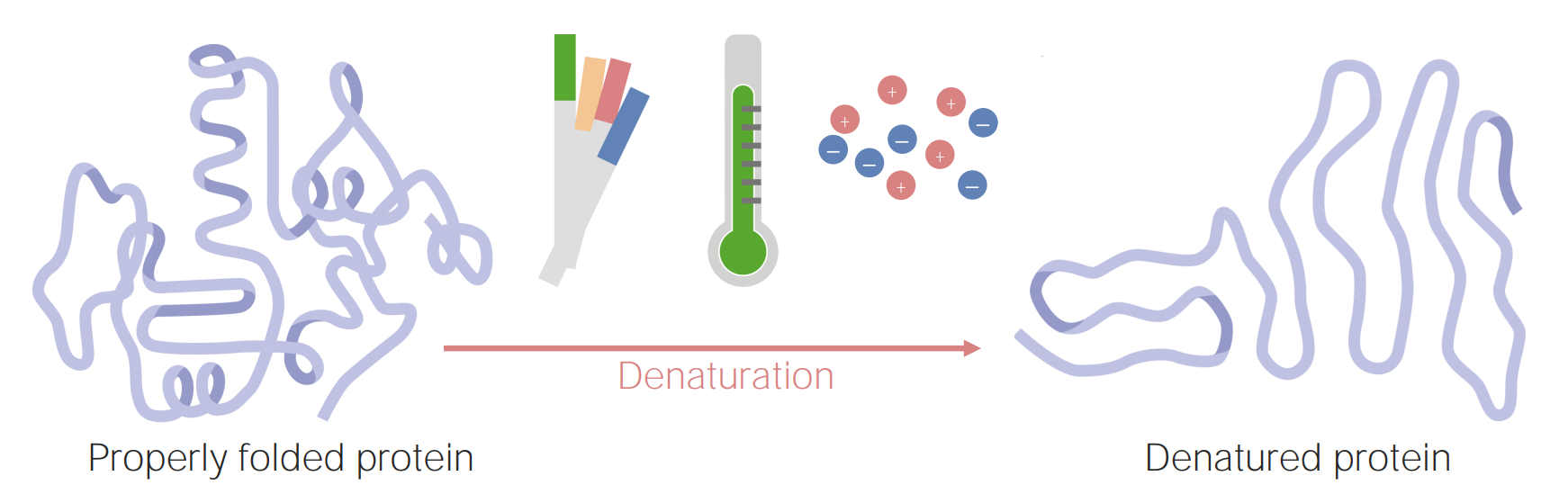
Proteins can become denatured (or unfolded) as a result of changes in pH, temperature, or ionic concentration.
Image by Lecturio.A protein’s unique structure (primary, secondary, tertiary, and quaternary) will give it physical and chemical properties that are important for the protein’s function. Some of these properties include:
Proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis have an extensive range of functions in the body, including:
| Enzyme | Zymogen (precursor) | Activated by | Notes on activity |
|---|---|---|---|
| Gastric enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes secreted into the stomach Stomach The stomach is a muscular sac in the upper left portion of the abdomen that plays a critical role in digestion. The stomach develops from the foregut and connects the esophagus with the duodenum. Structurally, the stomach is C-shaped and forms a greater and lesser curvature and is divided grossly into regions: the cardia, fundus, body, and pylorus. Stomach: Anatomy | |||
| Pepsin | Pepsinogen | Hydrochloric acid Hydrochloric acid A strong corrosive acid that is commonly used as a laboratory reagent. It is formed by dissolving hydrogen chloride in water. Gastric acid is the hydrochloric acid component of gastric juice. Caustic Ingestion (Cleaning Products) | Most efficient between hydrophobic AAs |
| Pancreatic enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes secreted into the duodenum Duodenum The shortest and widest portion of the small intestine adjacent to the pylorus of the stomach. It is named for having the length equal to about the width of 12 fingers. Small Intestine: Anatomy | |||
| Trypsin | Trypsinogen Trypsinogen The inactive proenzyme of trypsin secreted by the pancreas, activated in the duodenum via cleavage by enteropeptidase. Pancreatic Parameters | Enteropeptidase Enteropeptidase A specialized proteolytic enzyme secreted by intestinal cells. It converts trypsinogen into its active form trypsin by removing the n-terminal peptide. Digestion and Absorption |
|
| Chymotrypsin Chymotrypsin A serine endopeptidase secreted by the pancreas as its zymogen, chymotrypsinogen and carried in the pancreatic juice to the duodenum where it is activated by trypsin. It selectively cleaves aromatic amino acids on the carboxyl side. Pancreatic Parameters | Chymotrypsinogen | Trypsin | Most efficient between hydrophobic AAs |
| Carboxypeptidase Carboxypeptidase Enzymes that act at a free c-terminus of a polypeptide to liberate a single amino acid residue. Pancreatic Parameters | Procarboxypeptidase | Trypsin |
|
| Elastase | Proelastase | Trypsin | Same as carboxypeptidase Carboxypeptidase Enzymes that act at a free c-terminus of a polypeptide to liberate a single amino acid residue. Pancreatic Parameters |
| Enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes bound to the brush border Brush border Tubular System of enterocytes in the small intestines | |||
| Aminopeptidase Aminopeptidase A subclass of exopeptidases that act on the free n terminus end of a polypeptide liberating a single amino acid residue. Digestion and Absorption | NA | NA | Breaks down small peptides from their amino end (i.e., N-terminus) |
| Dipeptidases | NA | NA | Breaks peptide bonds between 2 AAs → 2 single AAs |
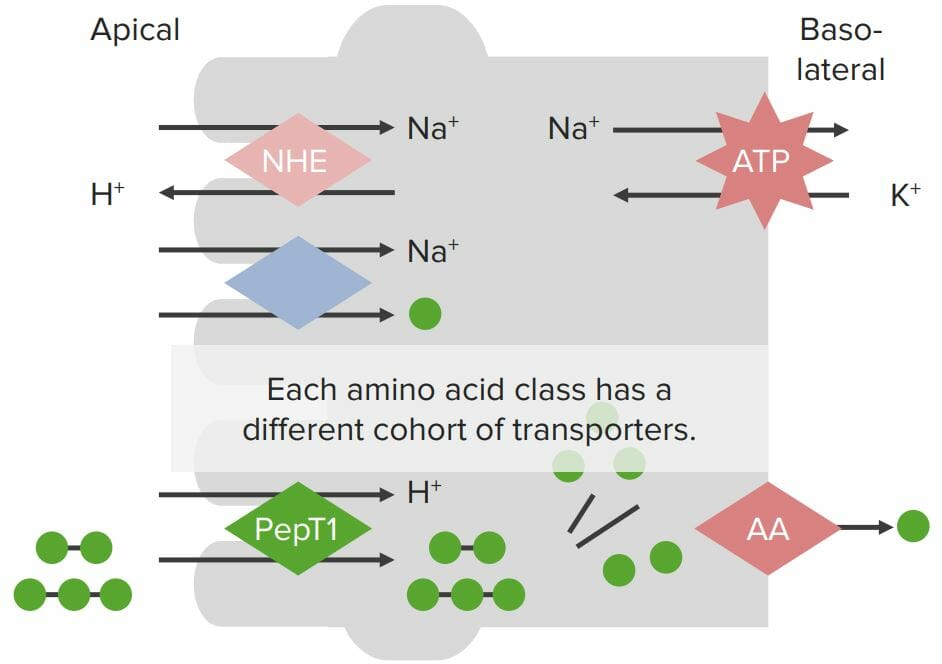
Transport proteins on enterocyte membranes involved in protein absorption:
The Na+/K+-ATPase on the basolateral membrane generates a Na+ gradient within the cell. A Na+/H+ exchanger (NHE) on the apical membrane also generates the H+ gradient. Individual amino acids (AAs; green balls) are absorbed via a Na+/AA cotransporter, where Na+ flows across the apical membrane into the enterocytes down its concentration gradient, bringing the AA with it (despite moving against the chemical AA gradient). Small peptides are absorbed via the H+/PepT cotransporter with H+ flowing down its concentration gradient into the cell, bringing the small peptides with it. Peptides are broken down into individual AAs by peptidases within the enterocytes. All AAs are then absorbed through specialized transporters on the basolateral membrane.
Protein metabolism refers to a group of biochemical processes responsible for both anabolism (the synthesis Synthesis Polymerase Chain Reaction (PCR) of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis and AAs) and catabolism (the breakdown of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis and AAs).
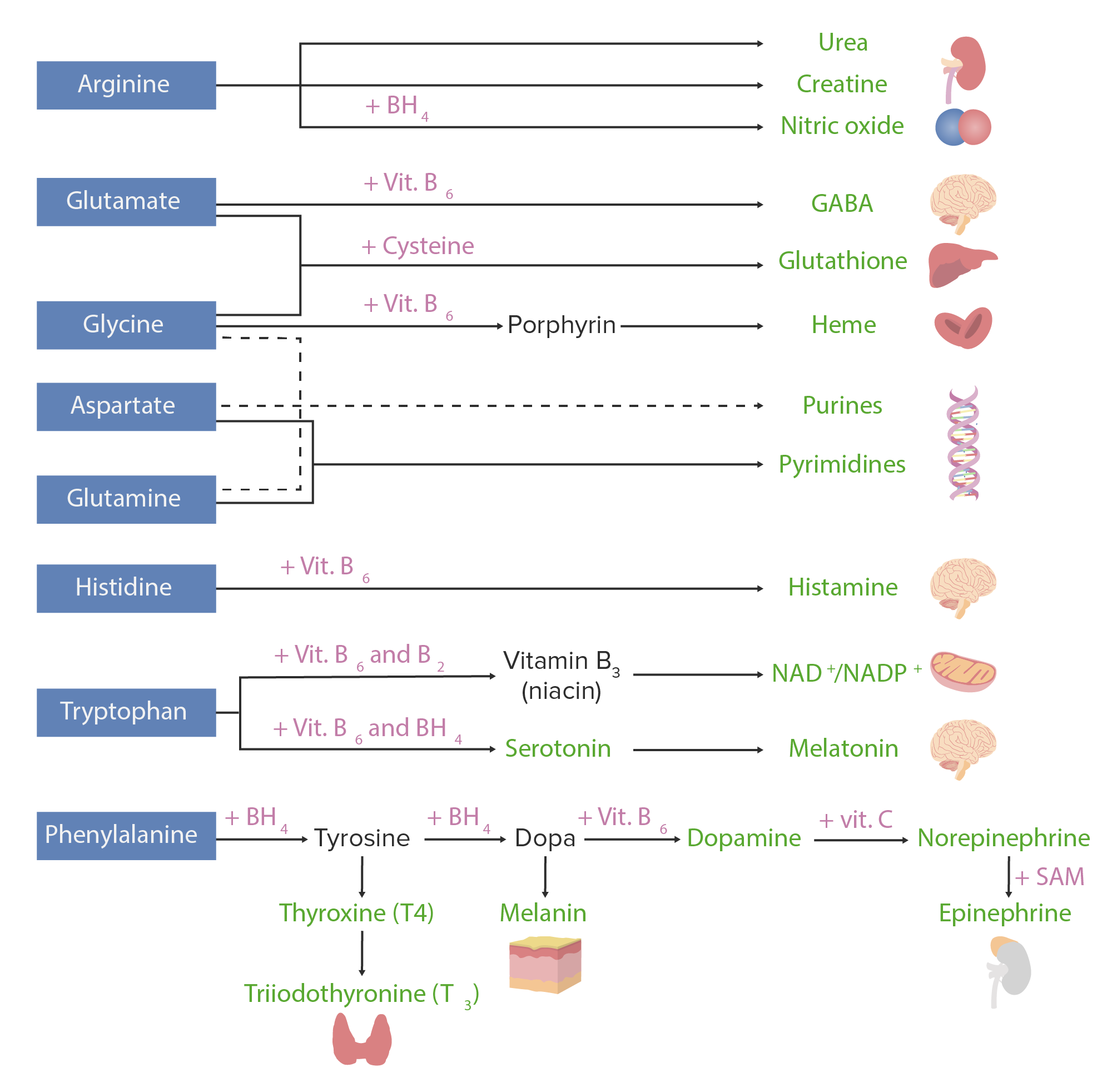
Amino acid derivatives:
Amino acids (in blue) are combined with certain cofactors or other substrates (in pink) to make several biologically-important substances (in green).
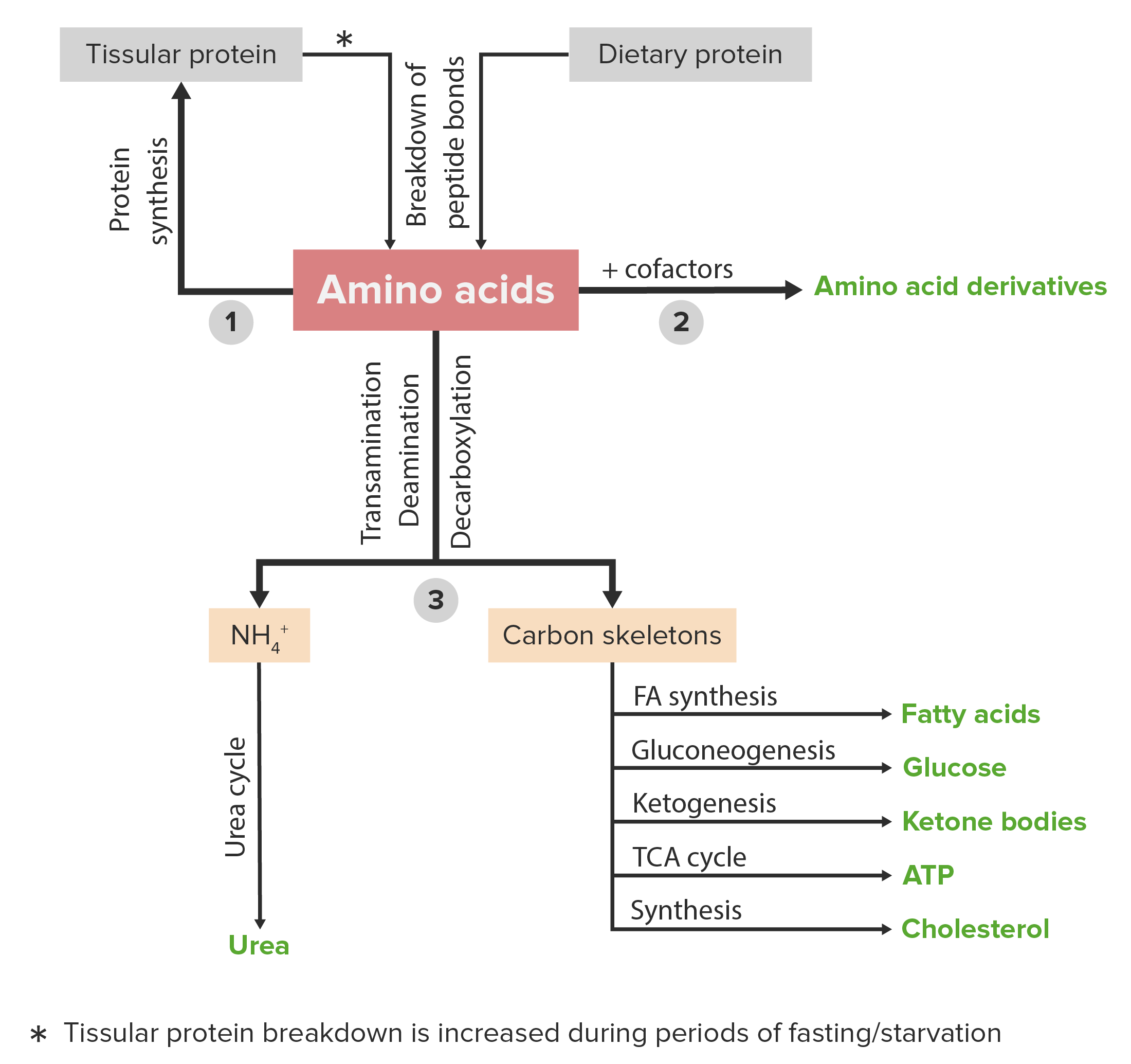
Schematic diagram of the metabolism of amino acids, including the 3 major pathways: reutilization in the synthesis of new proteins, union with cofactors to produce amino acid derivatives, and catabolism. Catabolism of amino acids includes the removal of functional groups and the breakdown of the carbon skeletons.
Image by Lecturio.A countless number of clinical disorders are caused by abnormalities or deficiencies of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis and/or abnormal protein metabolism. A few examples are listed below.