The cell undergoes a variety of changes in response to injury, which may or may not lead to cell death. Injurious stimuli trigger Trigger The type of signal that initiates the inspiratory phase by the ventilator Invasive Mechanical Ventilation the process of cellular adaptation Cellular adaptation In order to cope with their environment, cells undergo structural and functional changes. These cellular adaptations are reversible responses that allow cells to survive and continue to adequately function. Adaptive processes consist of increased cellular size and function (hypertrophy), increase in cell number (hyperplasia), decrease in cell size and metabolic activity (atrophy), or a change in the phenotype of the cells (metaplasia). Cellular Adaptation, whereby cells respond to withstand the harmful changes in their environment. Overwhelmed adaptive mechanisms lead to cell injury. Mild stimuli produce reversible injury Reversible Injury Ischemic Cell Damage. If the stimulus is severe or persistent, injury becomes irreversible. The principal targets of cell injury are the cell membranes, mitochondria Mitochondria Semiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes. The Cell: Organelles, protein synthesis Synthesis Polymerase Chain Reaction (PCR) machinery, and DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure. Multiple cellular abnormalities resulting from the damage result in cell death. The 2 main types of cell death are necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage and apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage. Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage is an uncontrolled cell death characterized by inflammatory changes in a pathologic condition. Apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage is programmed cell death, a mechanism with both physiologic and pathologic effects.
Last updated: Mar 30, 2026
Contents

Cellular response to stress and injury
Image by Lecturio.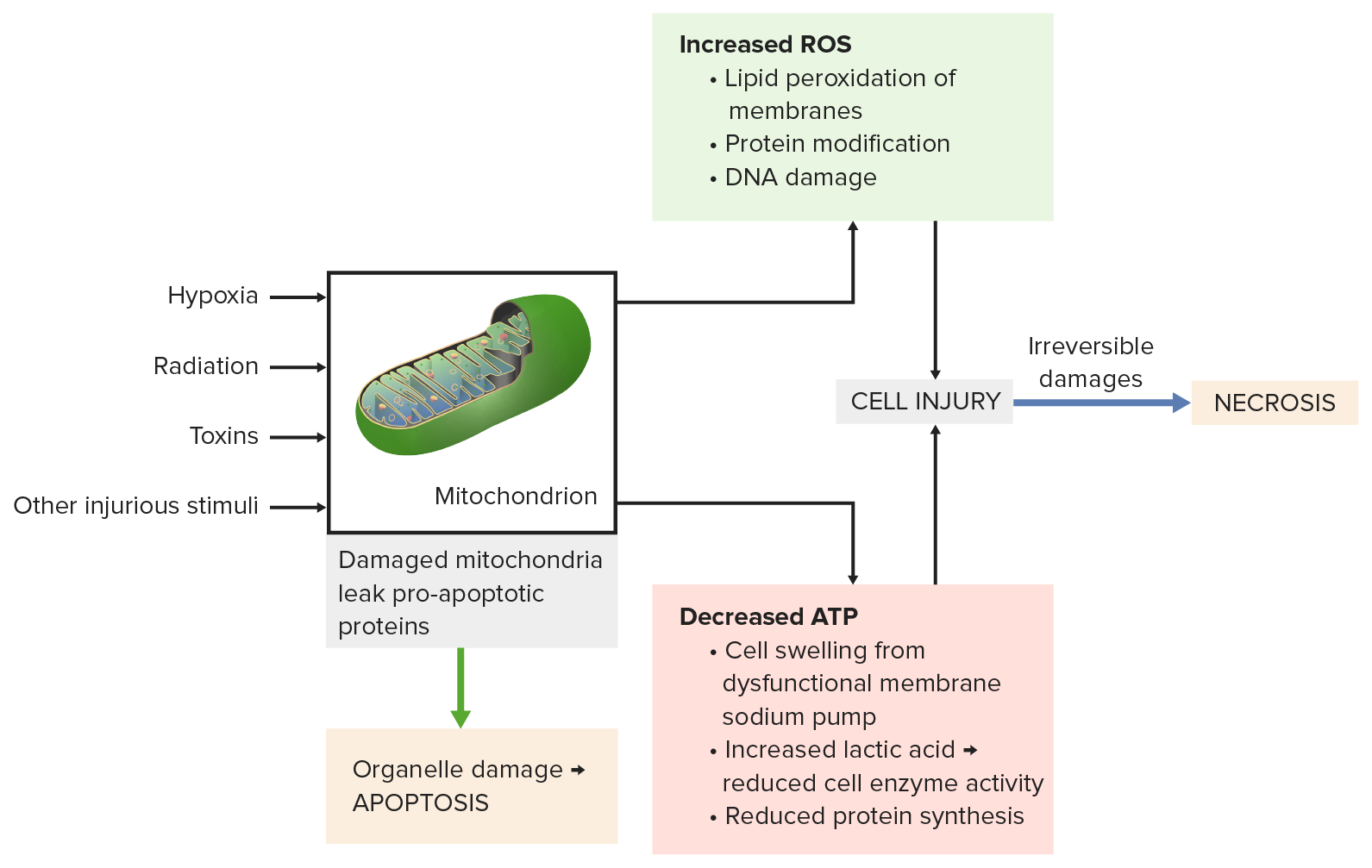
Mitochondrial damage from injurious stimuli (e.g., radiation, toxins) leads to:
Bottom left: Pro-apoptotic proteins leak from the mitochondria causing apoptosis.
Top right: Incomplete oxidative phosphorylation produces reactive oxygen species (ROS). Membranes, proteins and DNA are damaged.
Bottom right: Decreased ATP results in cell swelling, reduced enzyme activity and protein synthesis.
All processes lead to severe cell injury, then necrosis occurs.
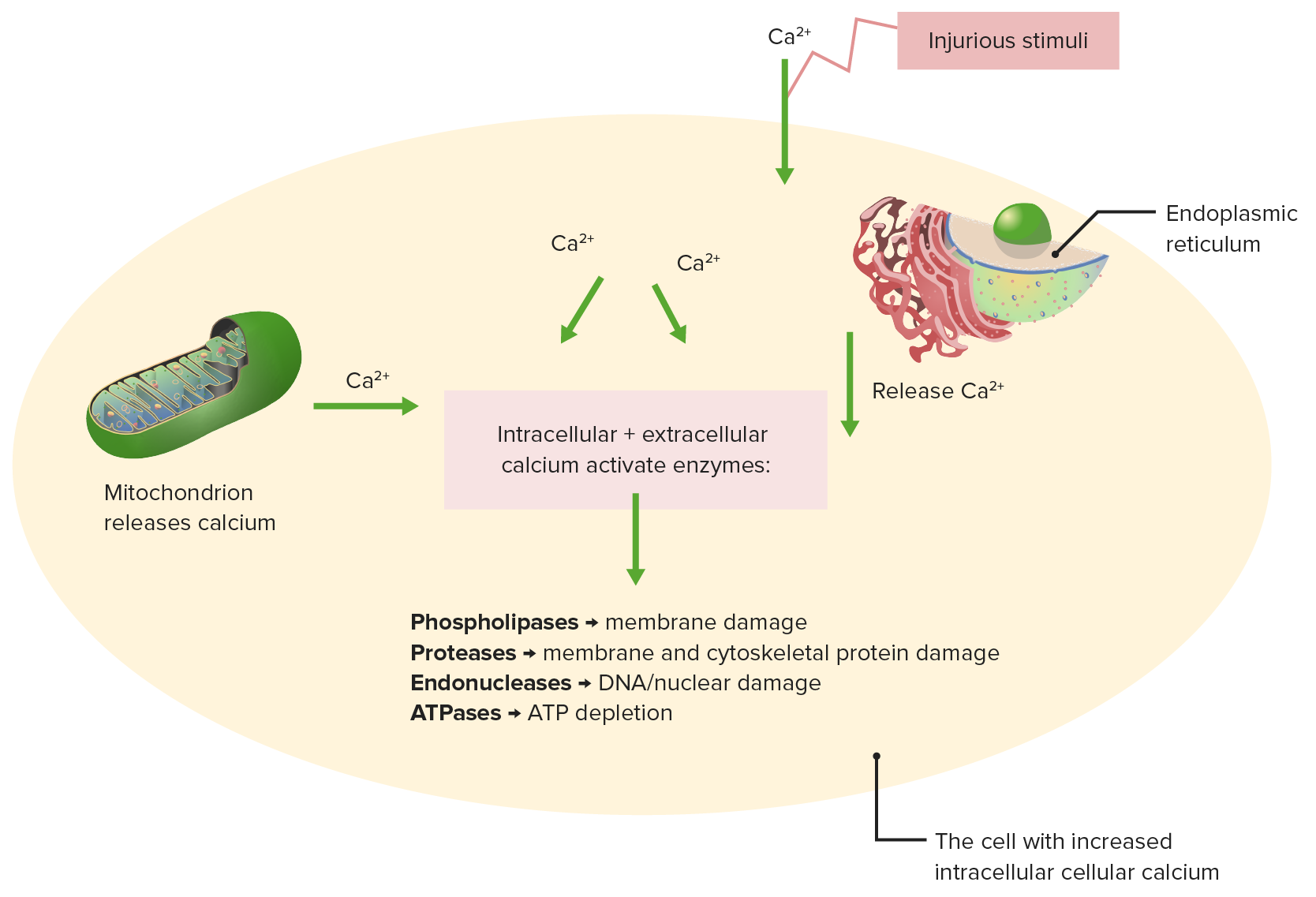
Effects of impaired calcium homeostasis
Injurious stimuli cause release of calcium from the mitochondrion and endoplasmic reticulum.
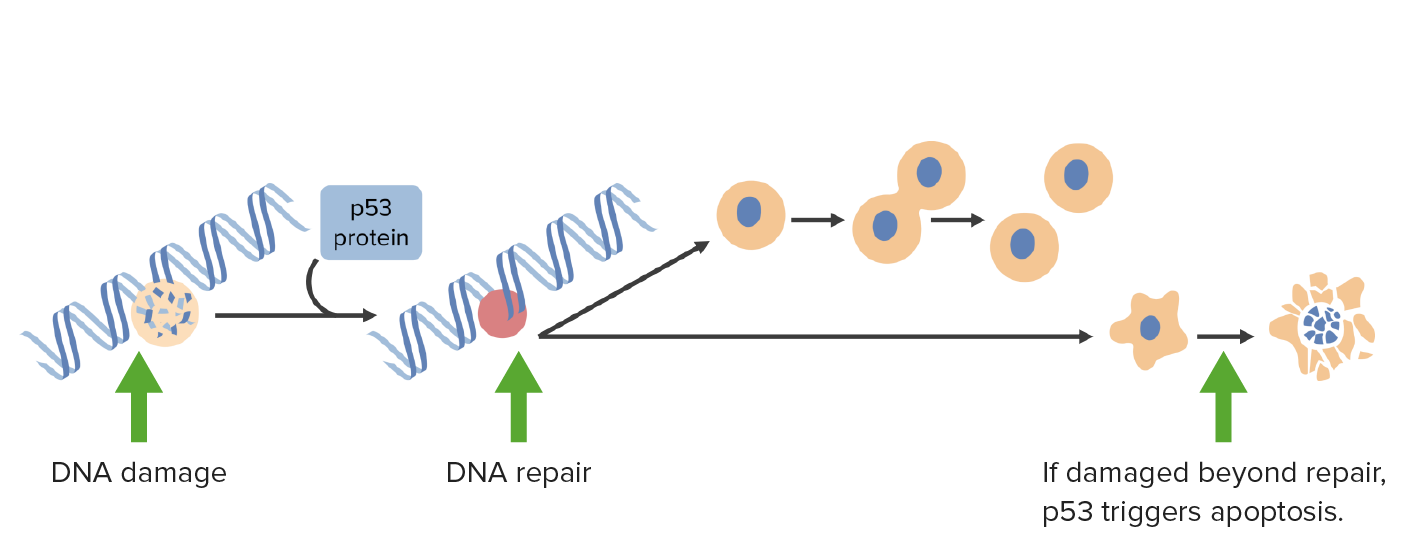
DNA damage activates p53 which arrests cells in G1 phase and triggers DNA repair mechanisms. If damage is irreparable, p53 triggers apoptosis.
Image by Lecturio.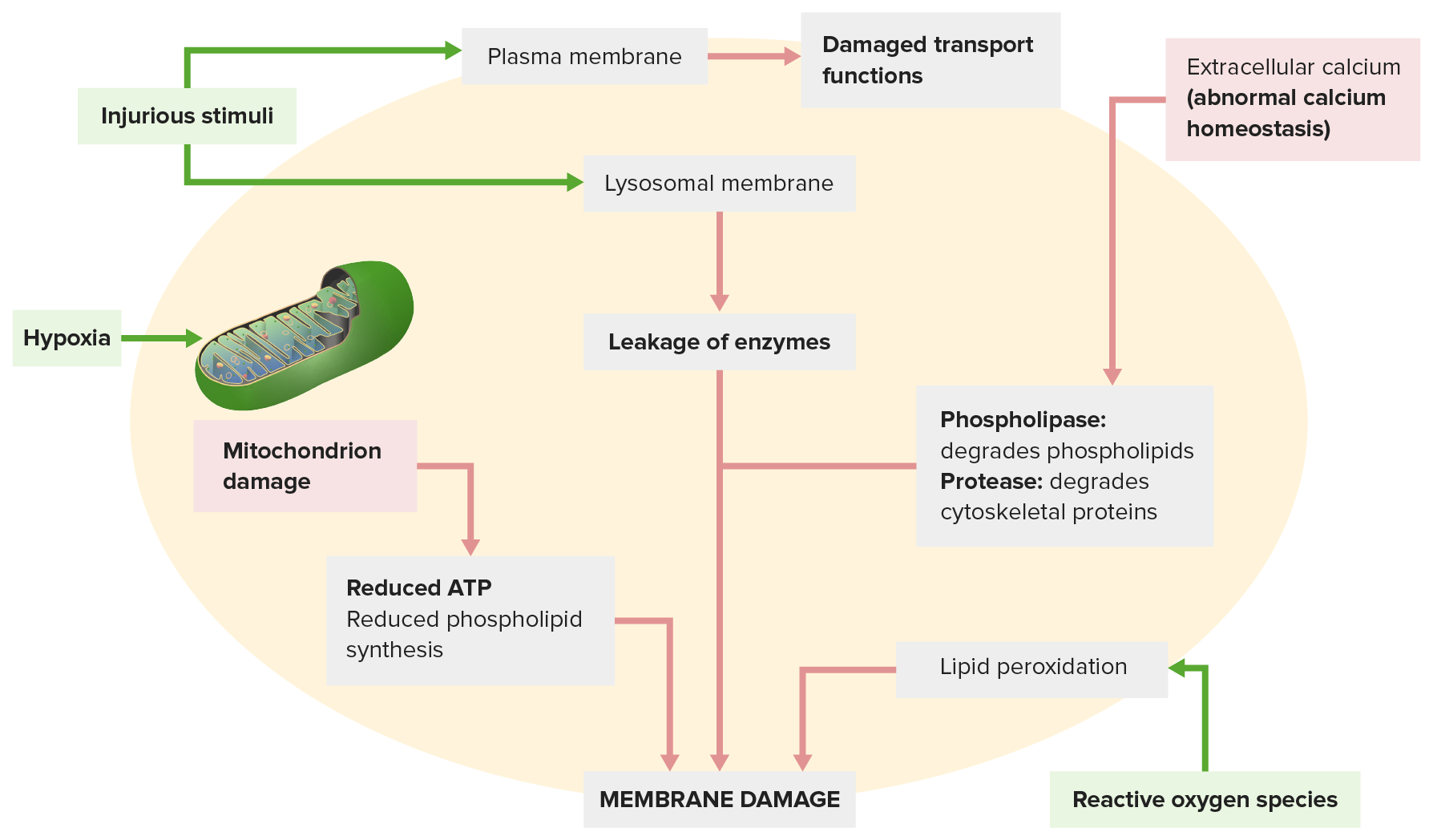
Membrane damage occurs from the following:
An injurious stimulus (top left) leads to disrupted transport functions. The injurious stimulus also affects lysosomal membranes, leaking enzymes that damage the cell.
Other mechanisms: Abnormal calcium homeostasis (top right) releases enzymes that degrade the membrane; mitochondrial dysfunction (lower left) reduces ATP production needed for membrane synthesis.
Reactive oxygen species (lower right) cause lipid peroxidation, leading to membrane phospholipid loss.
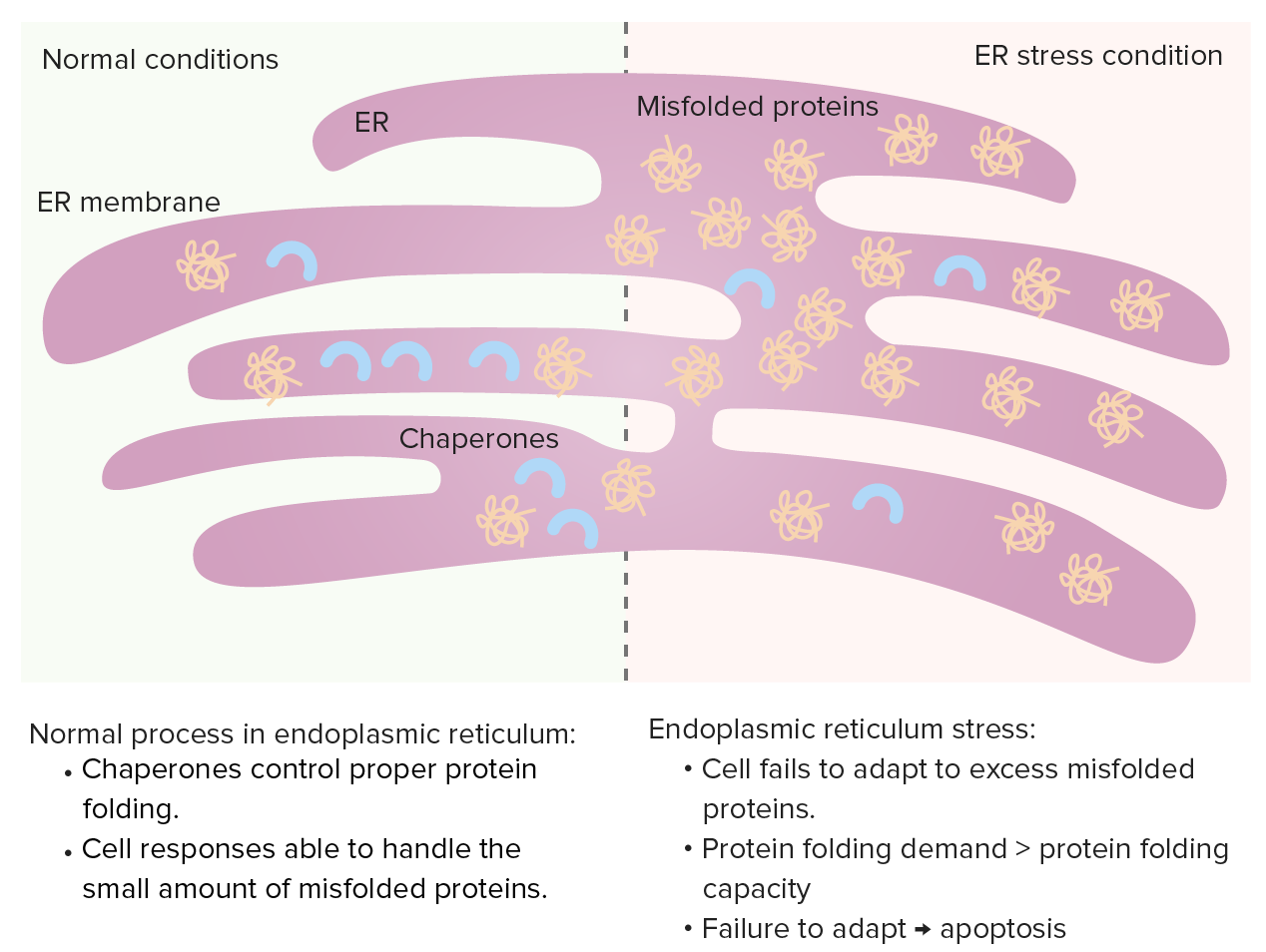
Endoplasmic reticulum (ER)
Chaperones control protein folding in the ER and misfolded proteins normally undergo proteolysis. When misfolded proteins increase, unfolded protein response occurs (increasing chaperones, decreasing protein synthesis and enhancing degradation of misfolded proteins).
ER stress: If protein folding demand increases (excessive misfolded proteins), the protein folding capacity gets saturated, leading to cell apoptosis.
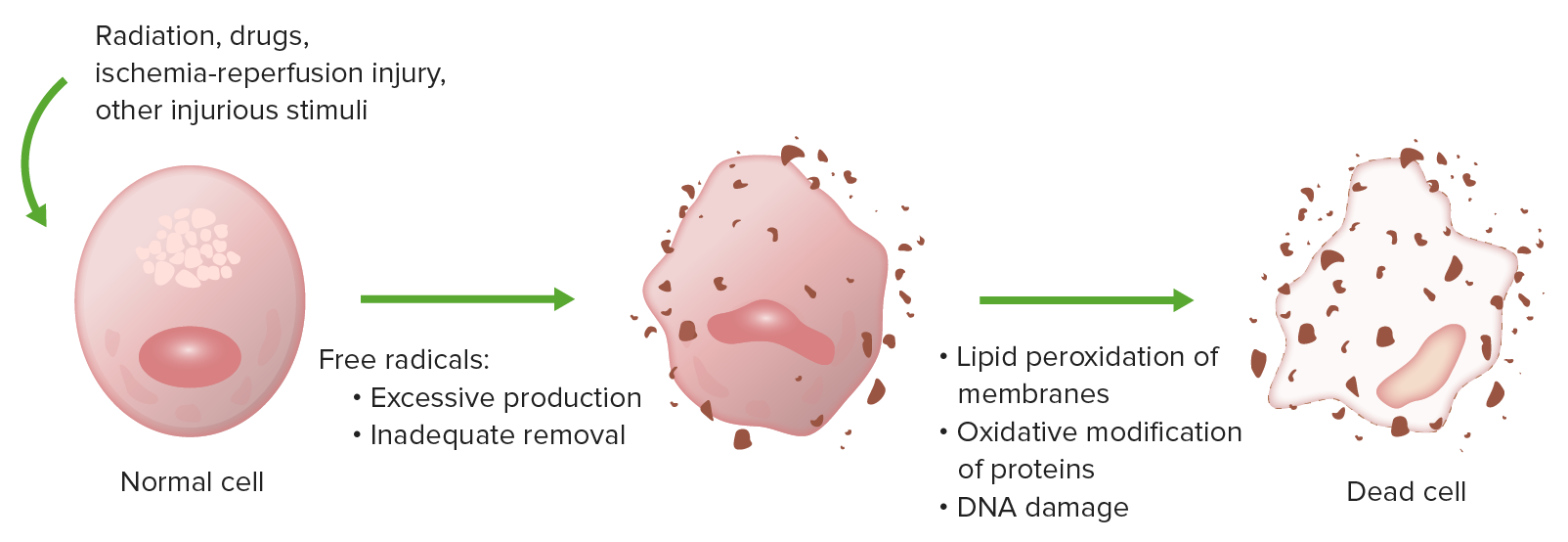
Oxidative stress causes cell injury by lipid peroxidation of membranes, oxidative modification of proteins, and DNA damage.
Image by Lecturio.Caspases:
Intrinsic pathway Intrinsic pathway The intrinsic pathway is mainly responsible for the amplification of factor X activation Hemostasis (initiation):
Extrinsic pathway Extrinsic pathway The extrinsic pathway is the primary physiological mechanism by which clotting is initiated Hemostasis (initiation):
Execution phase:
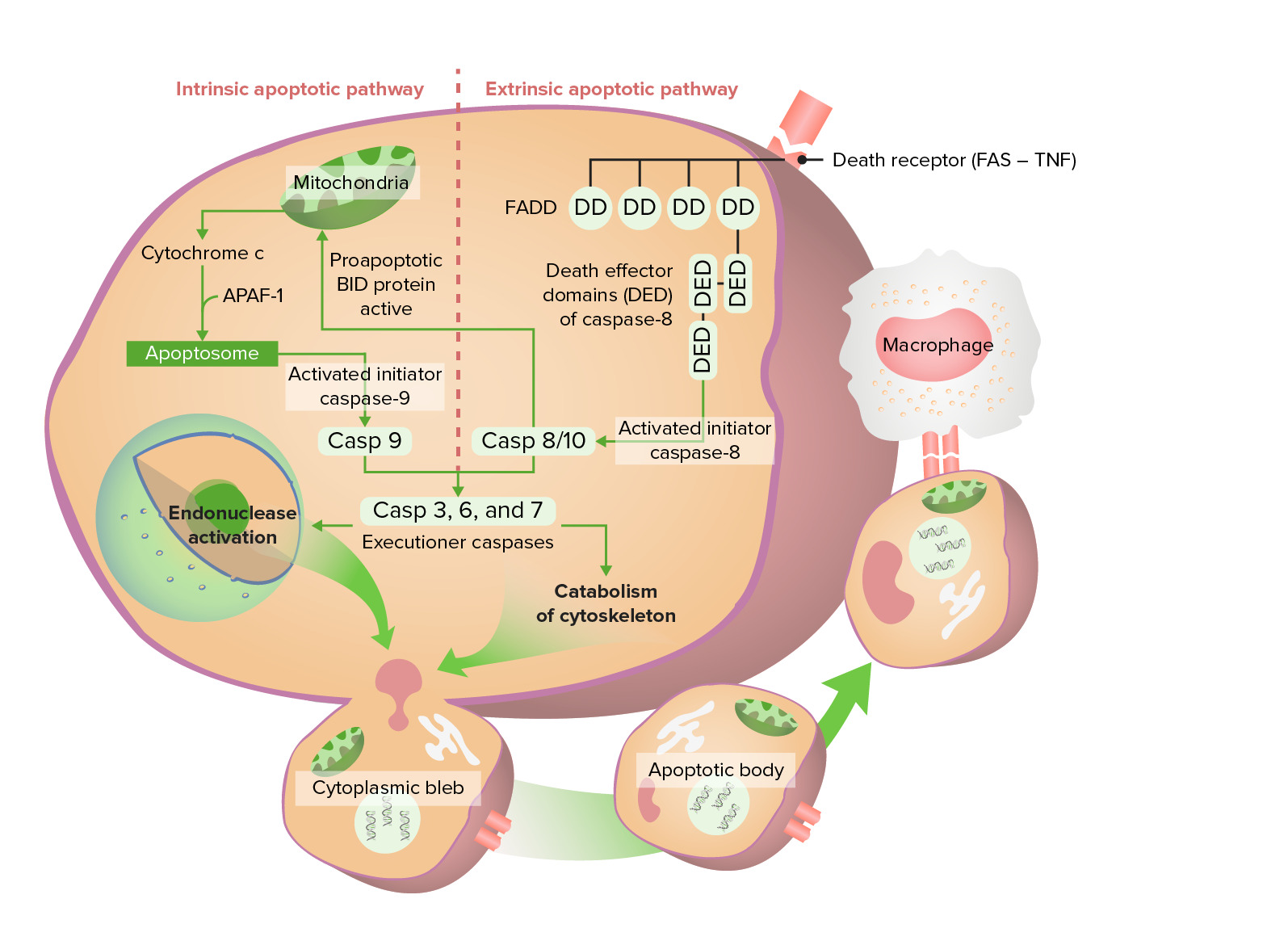
Intrinsic and extrinsic apoptotic pathway
Intrinsic pathway starts with release of cytochrome c, eventually activating caspase 9. The extrinsic pathway starts with activation of Fas (death receptor), which leads to an active caspase 8/10. These caspases go through the execution phase, finally forming apoptotic bodies which undergo phagocytosis.
| Features of necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage | Features of apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage | |
|---|---|---|
| Cell size | Enlarged ( swelling Swelling Inflammation) | Reduced (shrinkage) |
| Nucleus Nucleus Within a eukaryotic cell, a membrane-limited body which contains chromosomes and one or more nucleoli (cell nucleolus). The nuclear membrane consists of a double unit-type membrane which is perforated by a number of pores; the outermost membrane is continuous with the endoplasmic reticulum. A cell may contain more than one nucleus. The Cell: Organelles | Pyknosis Pyknosis Ischemic Cell Damage, karyorrhexis Karyorrhexis Ischemic Cell Damage, karyolysis Karyolysis Ischemic Cell Damage | Fragmentation Fragmentation Chronic Apophyseal Injury into nucleosome-size fragments |
| Plasma membrane Plasma membrane A cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules. The Cell: Cell Membrane | Disrupted | Intact but altered structure ( orientation Orientation Awareness of oneself in relation to time, place and person. Psychiatric Assessment of lipids Lipids Lipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes. Fatty Acids and Lipids) |
| Cellular contents | Enzymatic digestion Digestion Digestion refers to the process of the mechanical and chemical breakdown of food into smaller particles, which can then be absorbed and utilized by the body. Digestion and Absorption; leak out of the cell | Intact; released in apoptotic bodies |
| Adjacent inflammation Inflammation Inflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation | Frequent | No |
| Physiologic or pathologic role | Pathologic (result of irreversible cell injury) |
Physiologic:
elimination
Elimination
The initial damage and destruction of tumor cells by innate and adaptive immunity. Completion of the phase means no cancer growth.
Cancer Immunotherapy of unwanted cells Pathologic: cell injury from DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure and protein damage |
Coagulative necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage:
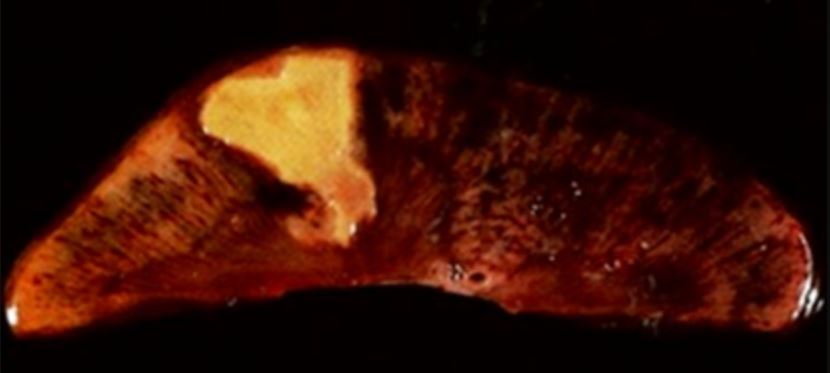
Coagulative necrosis in kidney tissue
Image: “Coagulative necrosis in kidney tissue” by Dentl college survival kit. License: Public DomainLiquefactive necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage:
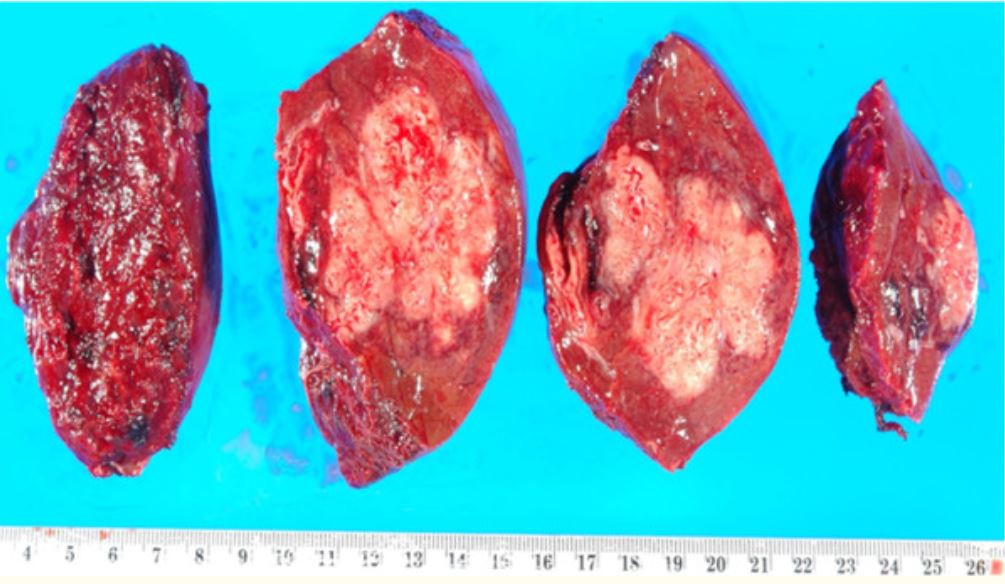
Resected portion of the liver with an abscess, which is a manifestation of liquefactive necrosis
Image: “Resection of a methicillin-resistant Staphylococcus aureus liver abscess in a patient with Crohn’s disease under infliximab treatment” by Togashi J, Sugawara Y, Akamatsu N, Aoki T, Ijichi M, Tanabe M, Kusaka K, Shibazaki M, Tadami T, Sakou M, Takazoe M, Bandai Y, Kokudo N . License: CCBY 2.0Caseous necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage :
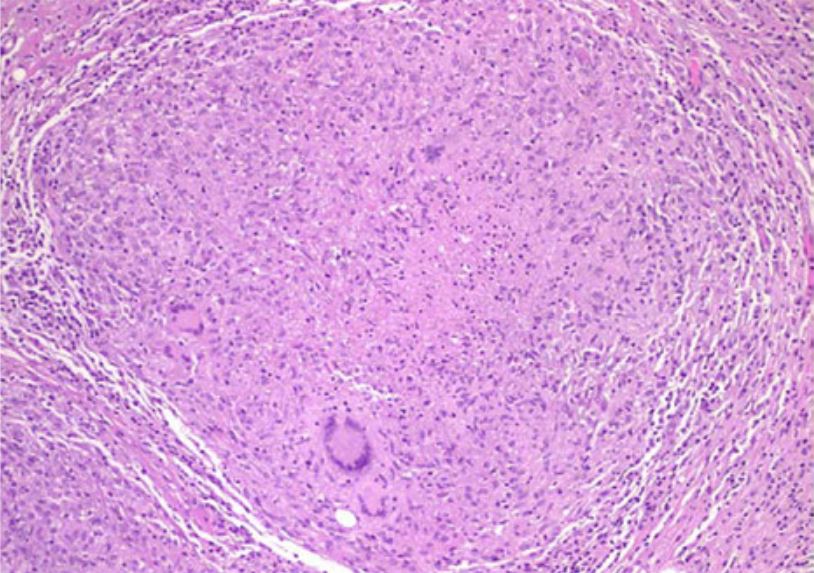
Histological examination demonstrating caseous necrotic granuloma in tuberculous peritonitis
Image: “Tuberculous peritonitis in pregnancy” by Lahbabi M, Brini J, Massaoudi K. License: CCBY 2.0Fat necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage:
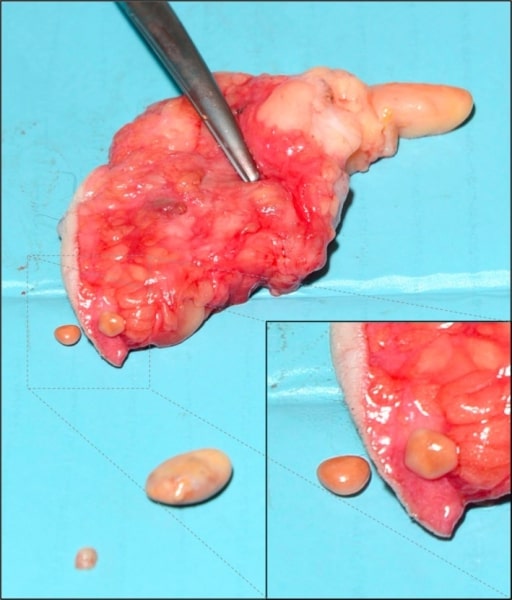
Subcutaneous fat necrosis of the breast: believed to be from trauma‐related ischemia and necrosis of fat tissue
Image: “Subcutaneous encapsulated fat necrosis” by Aydin D, Berg JO. License: CC BY 4.0Fibrinoid necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage:
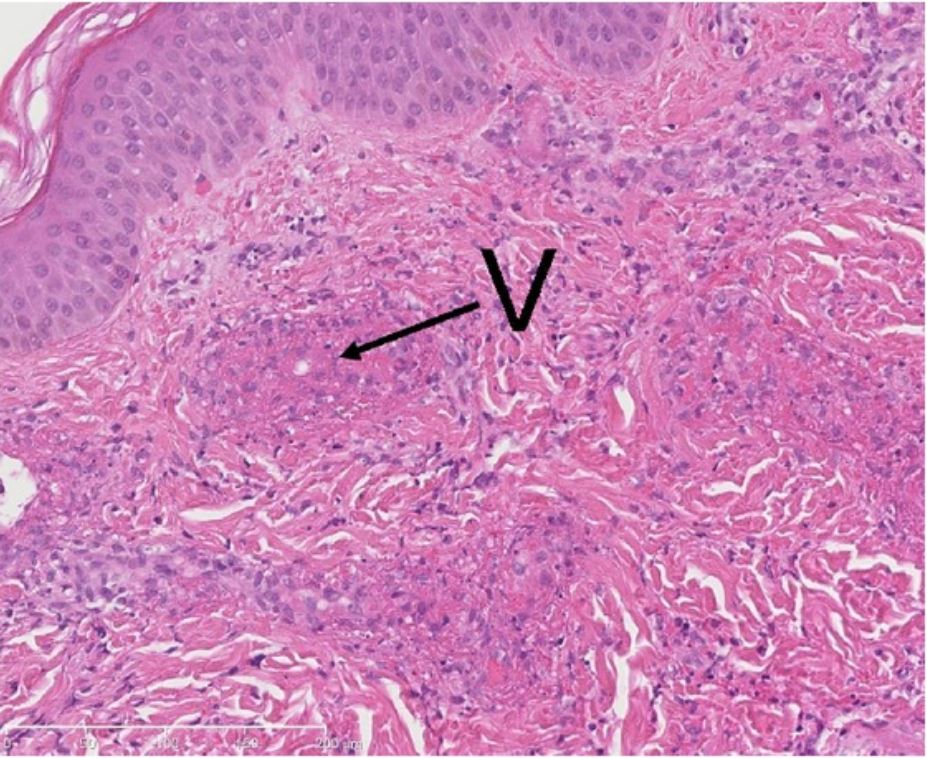
Biopsy from a cutaneous lesion (with leukocytoclastic vasculitis): a neutrophilic infiltrate and exudation of fibrin (fibrinoid necrosis) in the walls of small vessels (V).
Image: “Methylprednisolone therapy in acute hemorrhagic edema of infancy” by Risikesan J, Koppelhus U, Steiniche T, Deleuran M, Herlin T. License: CC BY 3.0Gangrenous necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage:

Image of a gangrenous foot: Affected toes were lost due to loss of blood supply.
Image: “Gangrene Foot” by آرمین. License: CC0 0.1Dystrophic calcification Dystrophic Calcification Cellular Accumulations:
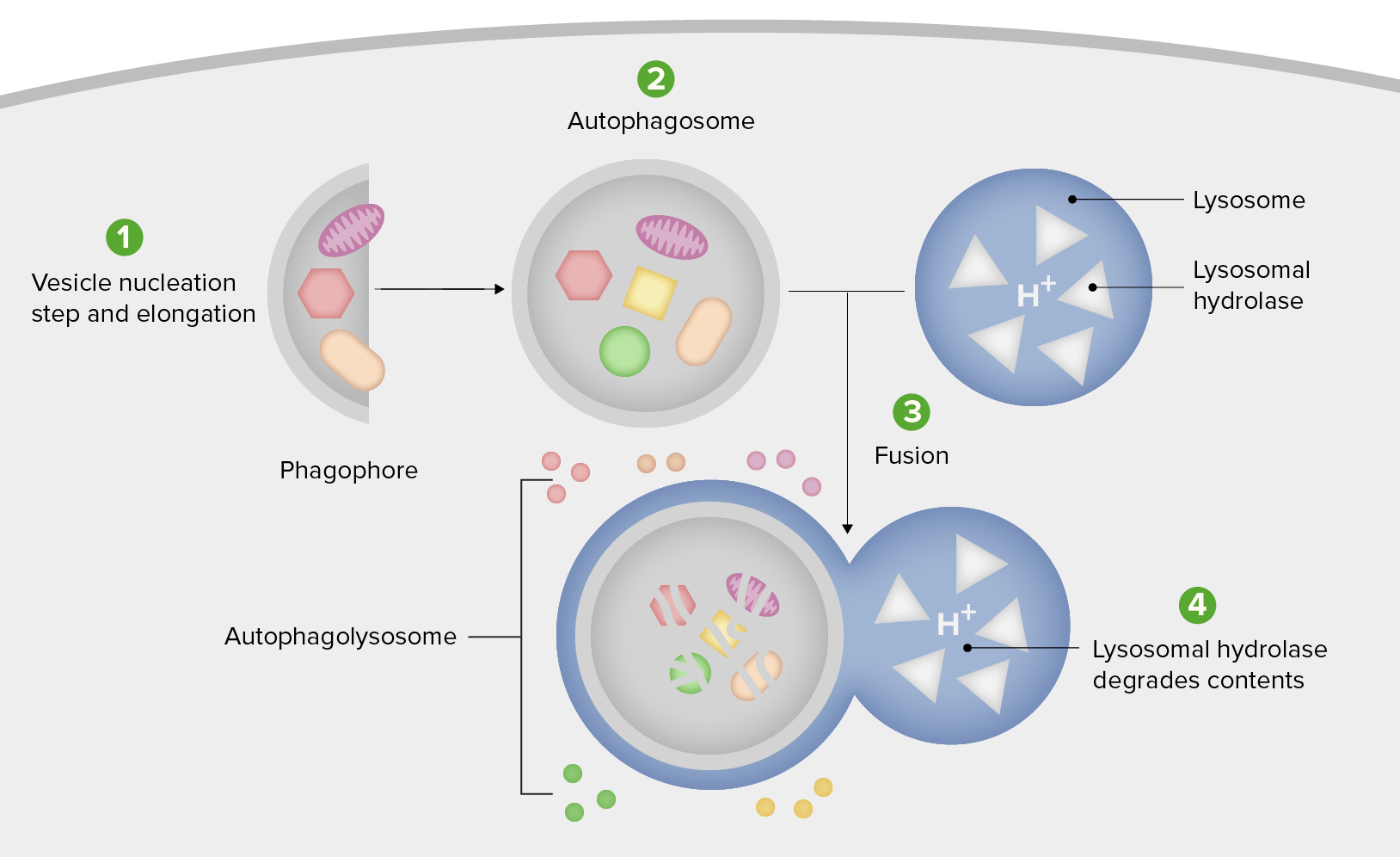
Schematic diagram of the steps of autophagy
1. Formation of the phagophore or isolation membrane (vesicle nucleation and elongation step).
2. Expansion of the phagophore into an autophagosome.
3. Fusion of the autophagosome with a lysosome forming an autophagolysosome.
4. Sequestered material is degraded inside the autophagolyosome and recycled.