La célula sufre una variedad de cambios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum respuesta a una lesión, que pueden conducir o no a la muerte celular. Los LOS Neisseria estímulos nocivos desencadenan el proceso de adaptación celular, mediante el cual las células responden para resistir los LOS Neisseria cambios dañinos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum su entorno. Los LOS Neisseria mecanismos adaptativos saturados conducen a una lesión celular. Los LOS Neisseria estímulos leves producen una lesión reversible. Si el estímulo es severo o persistente, la lesión se vuelve irreversible. Los LOS Neisseria principales objetivos de la lesión celular son las membranas celulares, las mitocondrias, la maquinaria de la síntesis proteica y el ADN. Múltiples anomalías celulares resultantes del daño provocan la muerte celular. Los LOS Neisseria 2 tipos principales de muerte celular son la necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage y la apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage. La necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage es una muerte celular descontrolada caracterizada por cambios inflamatorios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum una condición patológica. La apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage es la muerte celular programada, un mecanismo con efectos fisiológicos y patológicos.
Last updated: Mar 30, 2026
Contents

Respuesta celular al estrés y las lesiones
Imagen por Lecturio.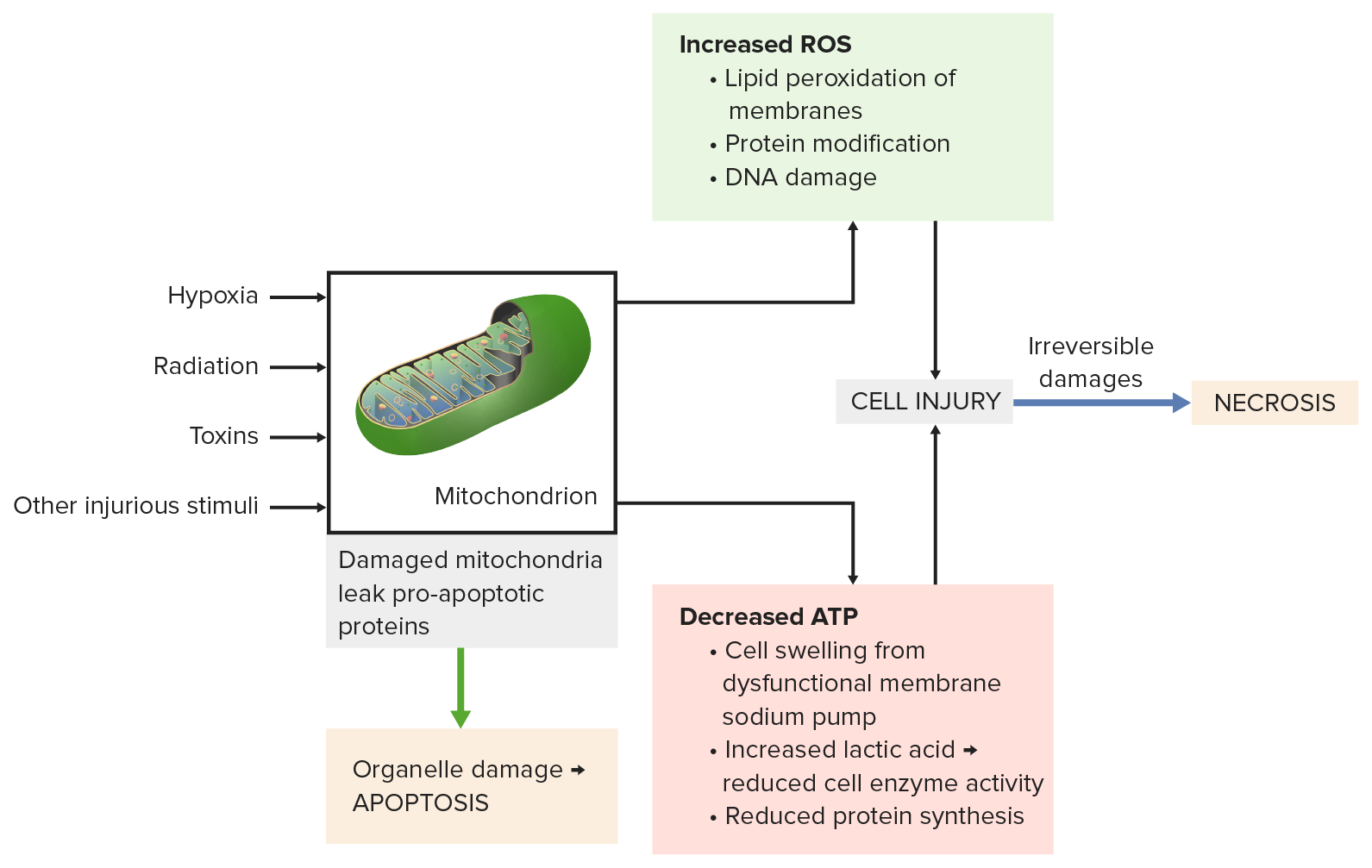
El daño mitocondrial por estímulos dañinos (e.g., radiación, toxinas) conduce a:
Abajo a la izquierda: las proteínas proapoptóticas se escapan de las mitocondrias y provocan la apoptosis.
Arriba a la derecha: la fosforilación oxidativa incompleta produce especies reactivas del oxígeno. Se dañan membranas, proteínas y ADN.
Abajo a la derecha: la disminución de ATP produce inflamación celular, reducción de la actividad enzimática y síntesis de proteínas.
Todos los procesos conducen a una lesión celular grave, luego se produce la necrosis.
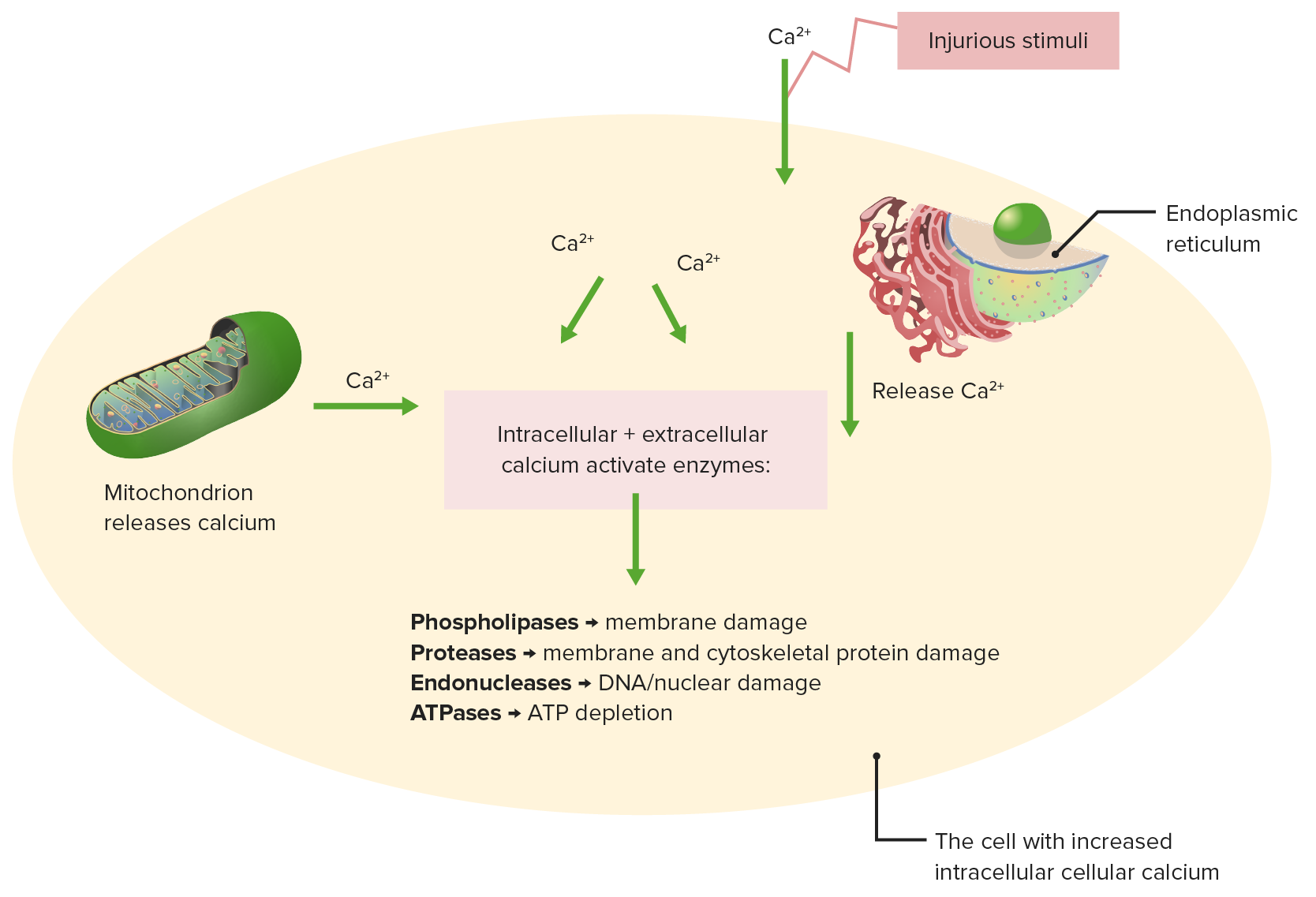
Efectos de la homeostasis del calcio alterada
Los estímulos nocivos provocan la liberación de calcio de la mitocondria y el retículo endoplásmico.
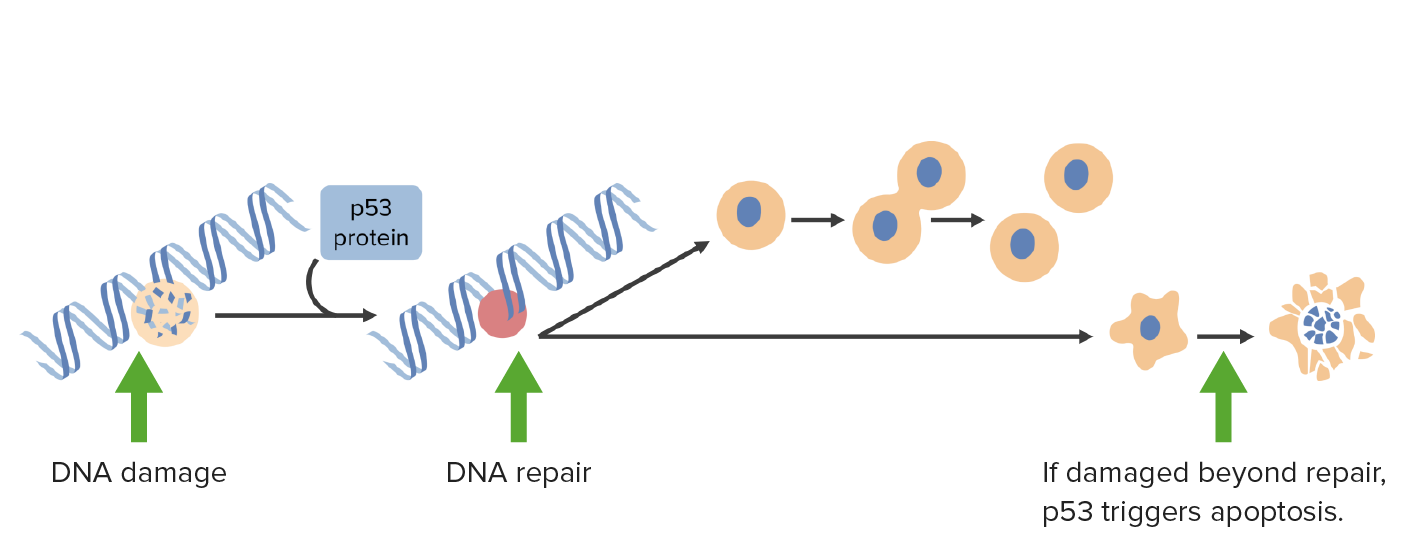
El daño del ADN activa la p53 que detiene las células en la fase G1 y desencadena los mecanismos de reparación del ADN. Si el daño es irreparable, la p53 activa la apoptosis.
Imagen por Lecturio.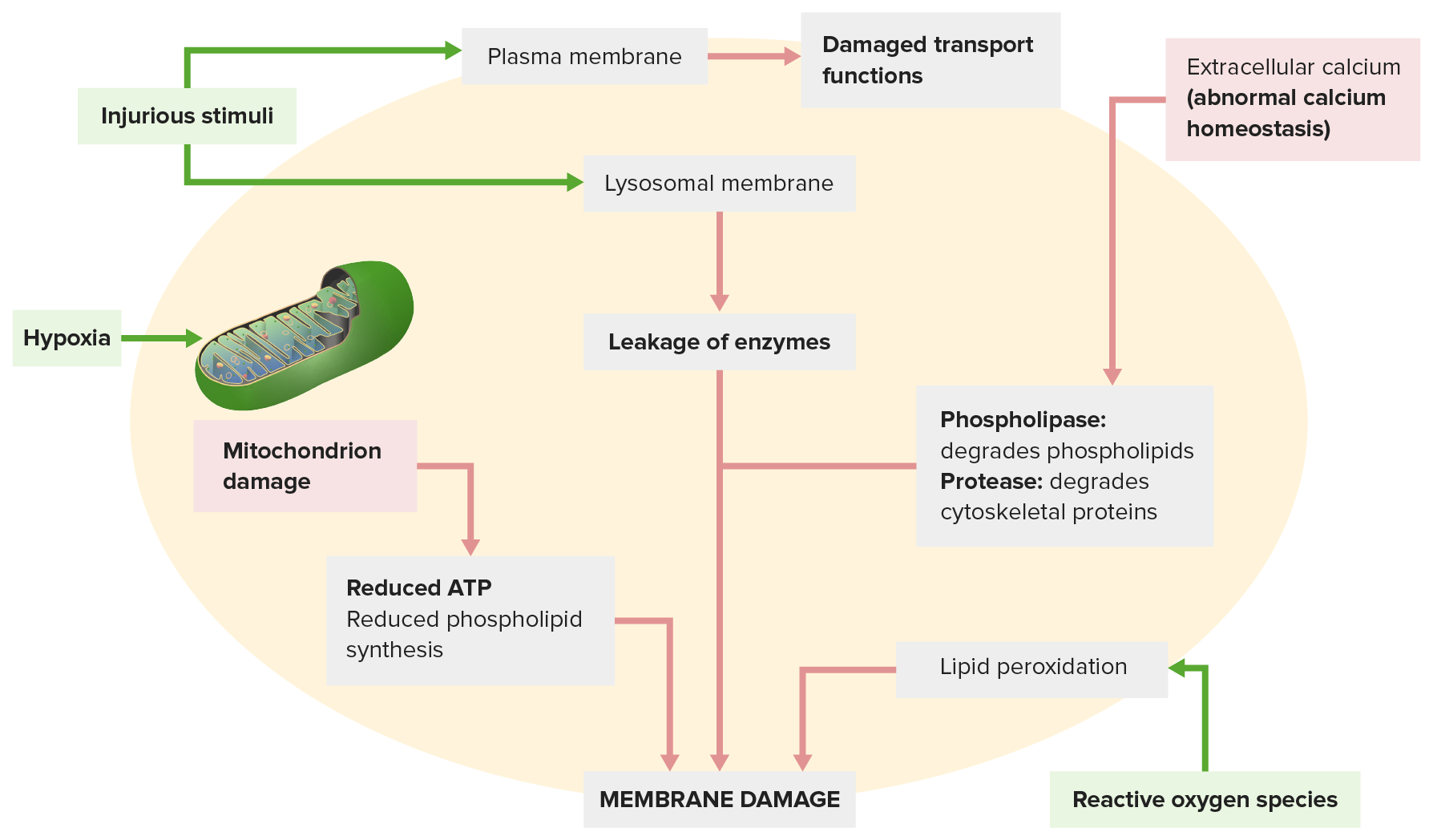
El daño de la membrana ocurre por lo siguiente:
Un estímulo dañino (arriba a la izquierda) conduce a la interrupción de las funciones de transporte. El estímulo dañino también afecta las membranas lisosomales, liberando enzimas que dañan la célula.
Otros mecanismos: la homeostasis anormal del calcio (arriba a la derecha) libera enzimas que degradan la membrana; la disfunción mitocondrial (abajo a la izquierda) reduce la producción de ATP necesaria para la síntesis de membrana.
Las especies reactivas de oxígeno (abajo a la derecha) causan peroxidación lipídica, lo que conduce a la pérdida de fosfolípidos de la membrana.
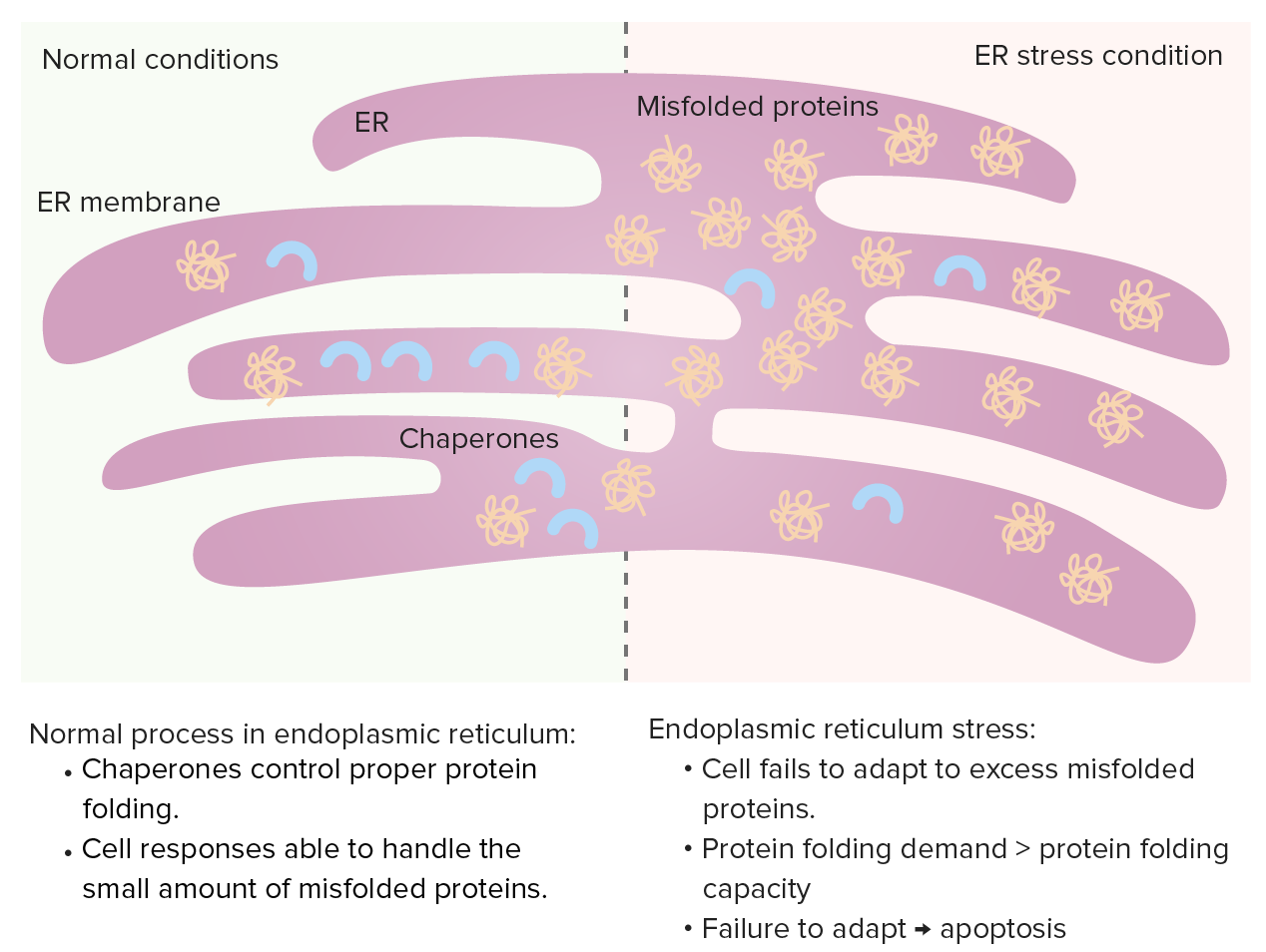
Retículo endoplásmico
Las chaperonas controlan el plegamiento de proteínas en el retículo endoplásmico y las proteínas mal plegadas normalmente se someten a proteólisis. Cuando aumentan las proteínas mal plegadas, se produce la respuesta a proteínas desplegadas (aumentando las chaperonas, disminuyendo la síntesis de proteínas y mejorando la degradación de las proteínas mal plegadas).
Estrés del retículo endoplásmico: si aumenta la demanda de plegamiento de proteínas (exceso de proteínas mal plegadas), la capacidad de plegado de proteínas se satura, lo que conduce a la apoptosis celular.
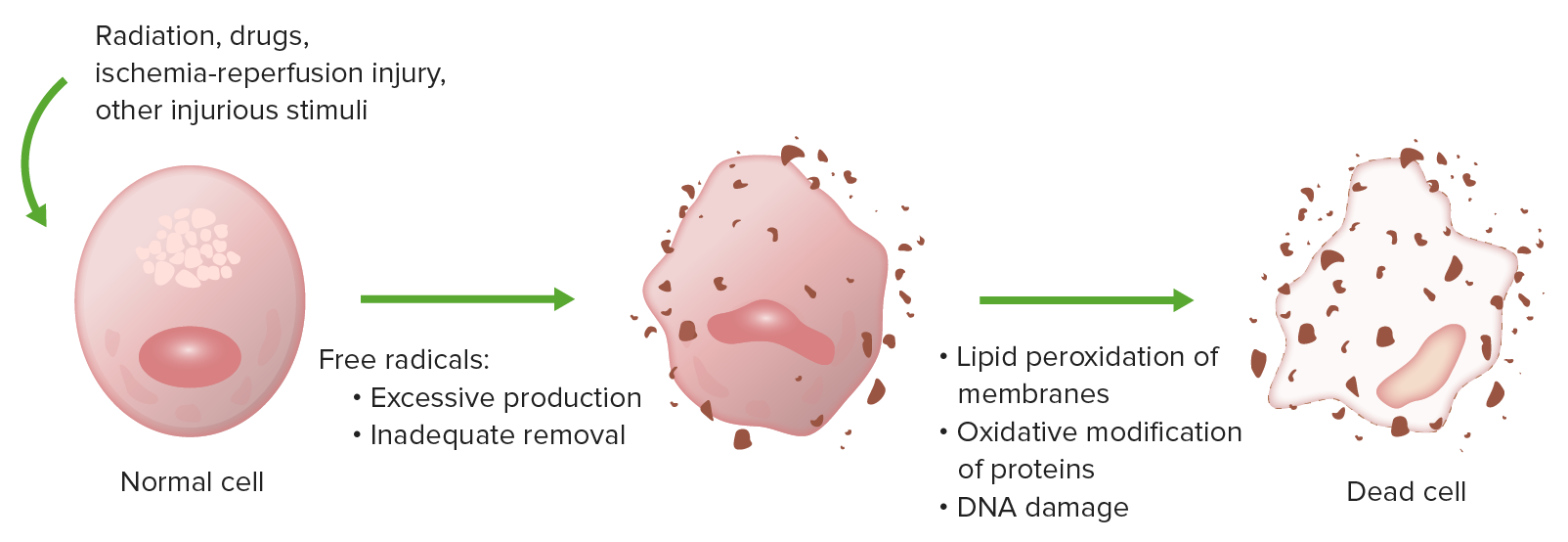
El estrés oxidativo causa lesión celular por peroxidación lipídica de membranas, modificación oxidativa de proteínas y daño al ADN.
Imagen por Lecturio.Caspasas:
Vía intrínseca (iniciación):
Vía extrínseca (iniciación):
Fase de ejecución:
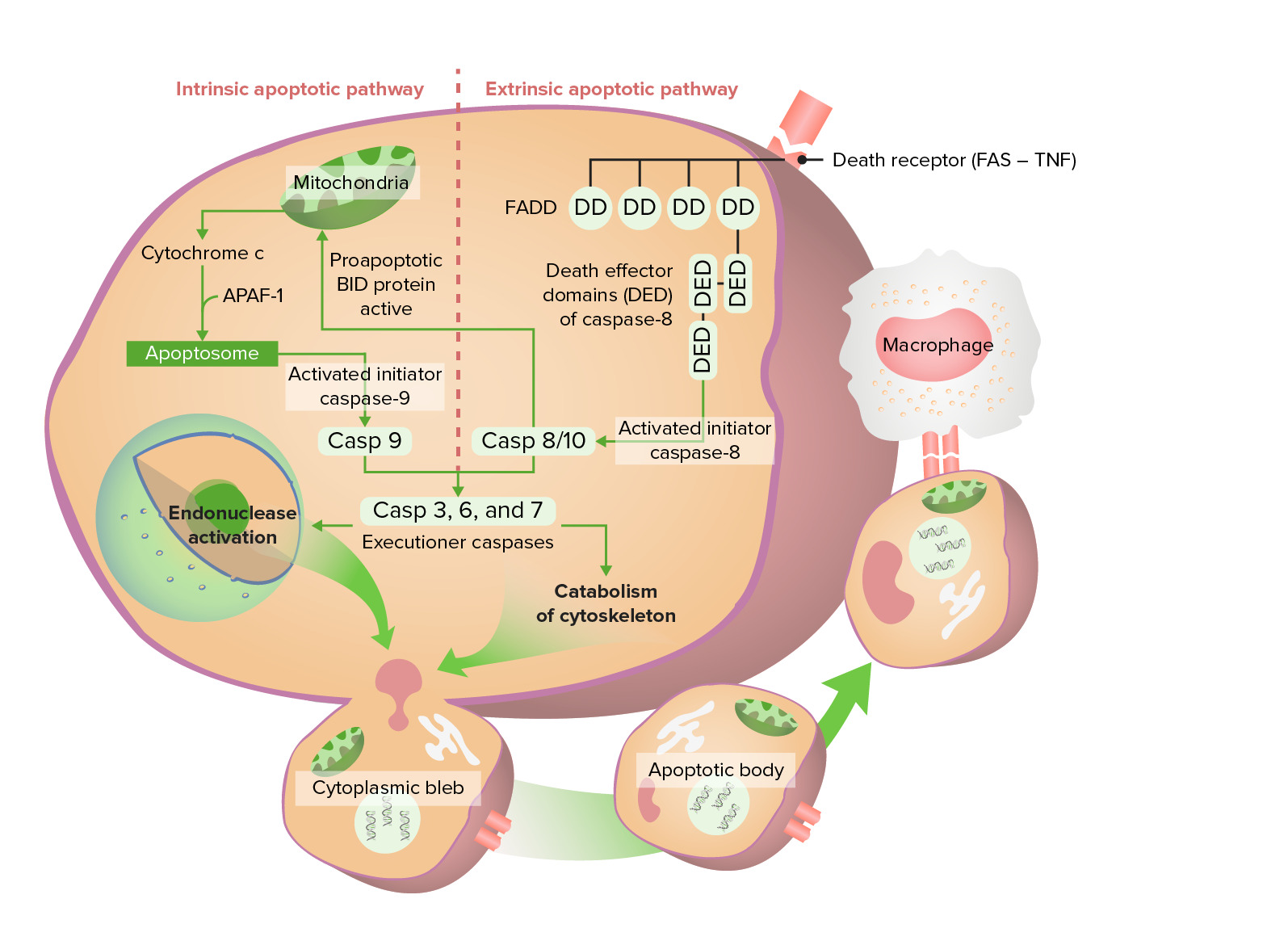
Vía apoptótica intrínseca y extrínseca
La vía intrínseca comienza con la liberación del citocromo c, que finalmente activa la caspasa 9. La vía extrínseca comienza con la activación de Fas (receptor de muerte), que conduce a la activación de caspasa 8/10. Estas caspasas pasan por la fase de ejecución, formando finalmente cuerpos apoptóticos que sufren fagocitosis.
| Características de la necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage | Características de la apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage | |
|---|---|---|
| Tamaño celular | Agrandada (edematizada) | Reducida (encogida) |
| Núcleo | Picnosis, cariorrexis, cariólisis | Fragmentación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum piezas del tamaño de un nucleosoma |
| Membrana plasmática | Dañada | Estructura intacta pero alterada (orientación de los LOS Neisseria lípidos) |
| Contenidos celulares | Digestión enzimática; fuga fuera de la célula | Intactos; liberados en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum cuerpos apoptóticos |
| Inflamación adyacente | Frecuente | No |
| Papel fisiológico o patológico | Patológico (resultado de lesión celular irreversible) |
Fisiológico: eliminación de células no deseadas. Patológico: lesión celular por daño del ADN y de las proteínas. |
Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage coagulativa :
Necrosis coagulativa en tejido renal
Imagen: “Coagulative necrosis in kidney tissue” por Dentl college survival kit. Licencia: Dominio PúblicoNecrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage licuefactiva:
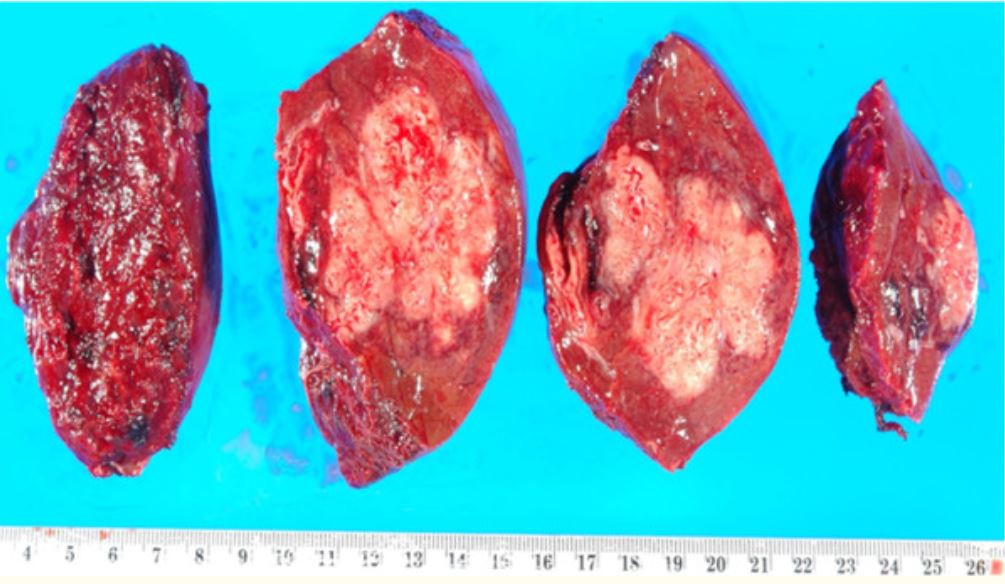
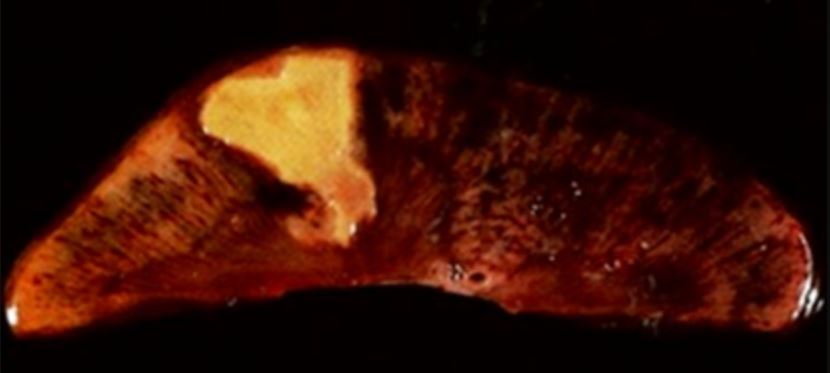
Porción resecada de un hígado con un absceso, el cual es una manifestación de necrosis licuefactiva
Imagen: “Resection of a methicillin-resistant Staphylococcus aureus liver abscess in a patient with Crohn’s disease under infliximab treatment” por Togashi J, Sugawara Y, Akamatsu N, Aoki T, Ijichi M, Tanabe M, Kusaka K, Shibazaki M, Tadami T, Sakou M, Takazoe M, Bandai Y, Kokudo N. Licencia: CCBY 2.0Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage caseosa:
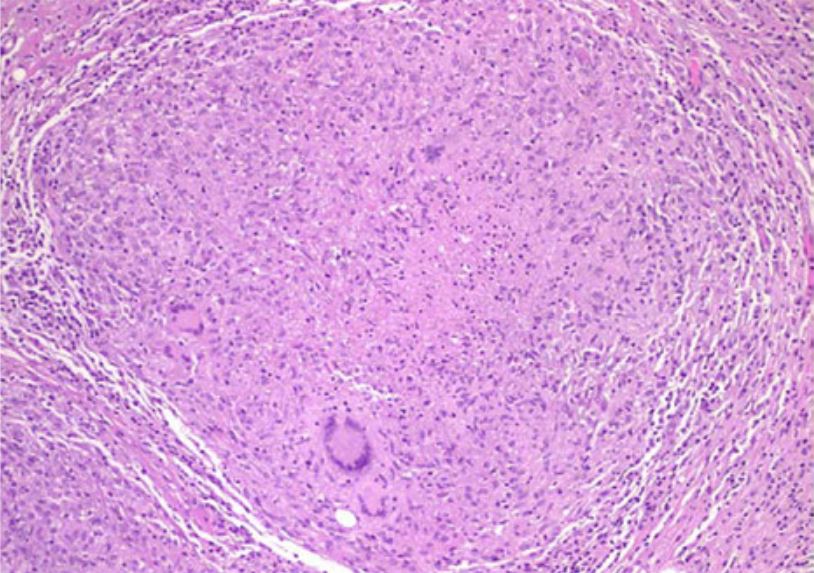
Examen histológico que muestra un granuloma necrótico caseoso en la peritonitis tuberculosa
Imagen: “Tuberculous peritonitis in pregnancy” por Lahbabi M, Brini J, Massaoudi K. Licencia: CCBY 2.0Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage grasa:
Necrosis grasa subcutánea de la mama: se cree que se debe a isquemia y necrosis del tejido graso, relacionados con traumatismo
Imagen: “Subcutaneous encapsulated fat necrosis” por Aydin D, Berg JO. Licencia: CC BY 4.0Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage fibrinoide:
Biopsia de lesión cutánea (con vasculitis leucocitoclástica): infiltrado neutrofílico y exudación de fibrina (necrosis fibrinoide) en las paredes de pequeños vasos (V).
Imagen: “Methylprednisolone therapy in acute hemorrhagic edema of infancy” por Risikesan J, Koppelhus U, Steiniche T, Deleuran M, Herlin T. License: CC BY 3.0Necrosis Necrosis The death of cells in an organ or tissue due to disease, injury or failure of the blood supply. Ischemic Cell Damage gangrenosa:

Imagen de un pie gangrenoso: los dedos afectados se perdieron debido a la pérdida de irrigación sanguínea.
Imagen: “Gangrene Foot” por آرمین. Licencia: CC0 0.1Calcificación distrófica:
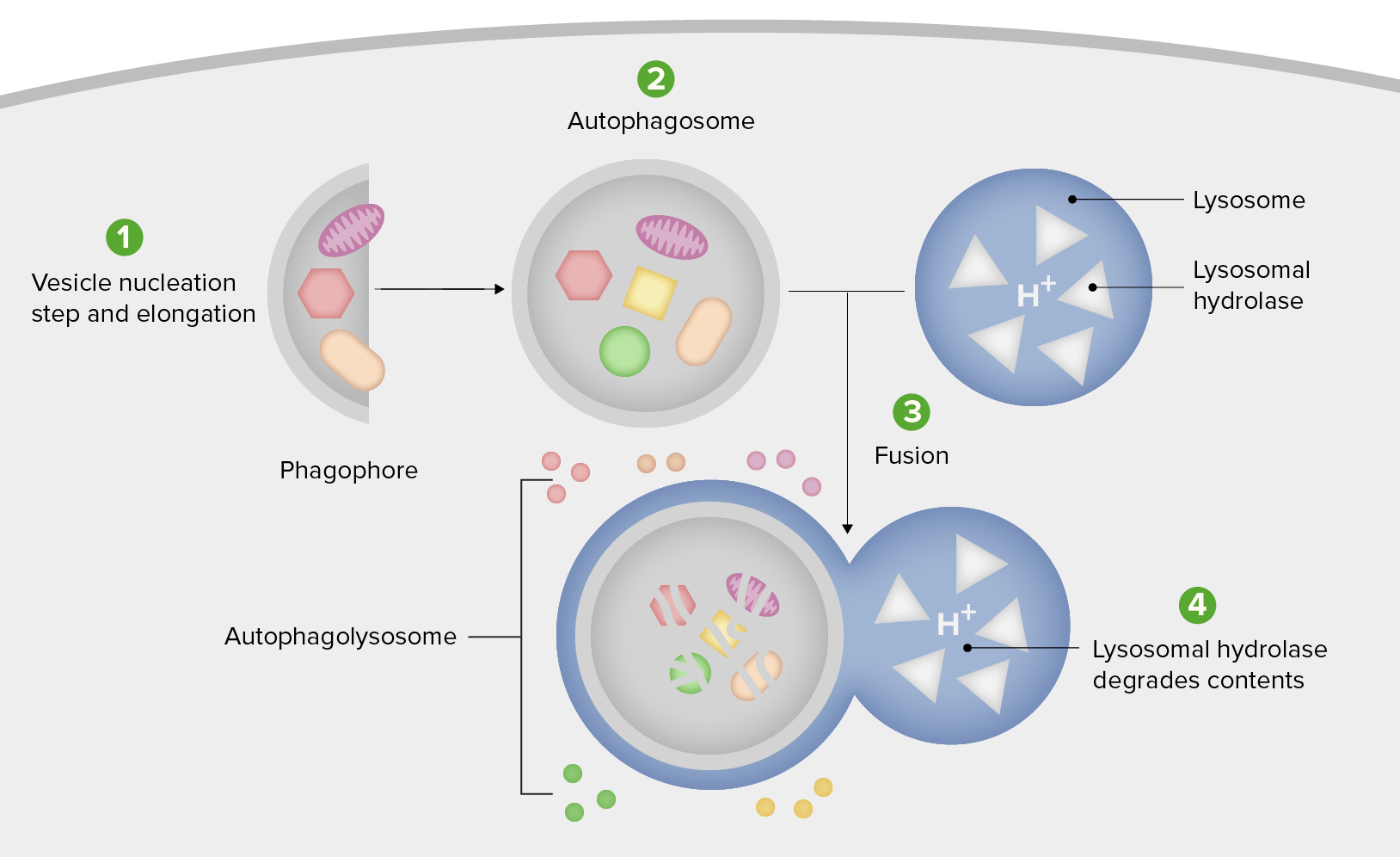
Diagrama esquemático de los pasos de la autofagia
1. Formación del fagóforo o membrana de aislamiento (etapa de nucleación y elongación de vesículas).
2. Expansión del fagóforo en un autofagosoma.
3. Fusión del autofagosoma con un lisosoma formando un autofagolisosoma.
4. El material secuestrado es degradado dentro del autofagoliosoma y reciclado.