Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other 'foreign' surfaces.Coagulation Studies, activation, and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies to the damaged vascular endotheliumEndotheliumA layer of epithelium that lines the heart, blood vessels (vascular endothelium), lymph vessels (lymphatic endothelium), and the serous cavities of the body.Arteries: Histology, forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade resulting in the formation of a more stable plug. Finally, as the vasculature is repaired, the clot is broken down in the fibrinolytic phaseFibrinolytic phaseCoagulation Studies.
Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation.
Phases of the hemostatic process
Constriction of the blood vessel: to limitLimitA value (e.g., pressure or time) that should not be exceeded and which is specified by the operator to protect the lungInvasive Mechanical Ventilationblood flowBlood flowBlood flow refers to the movement of a certain volume of blood through the vasculature over a given unit of time (e.g., mL per minute).Vascular Resistance, Flow, and Mean Arterial Pressure to the area
Formation of the platelet plug: the initial, temporary plug
Activation of the coagulation cascade: to form a more stable fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis clot
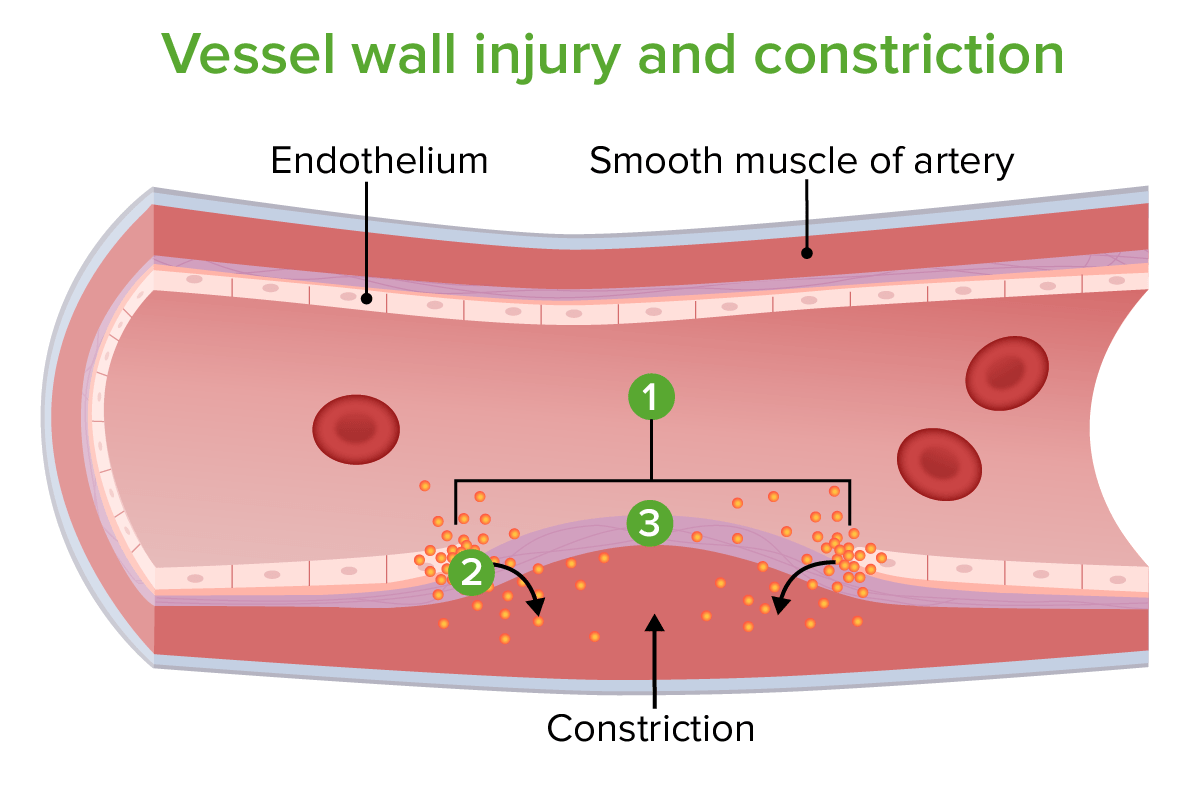
Vessel wall injury and constriction:
1. Site of injury
2. Constriction caused by endothelin release
3. Exposed collagen fibers
Image by Lecturio.
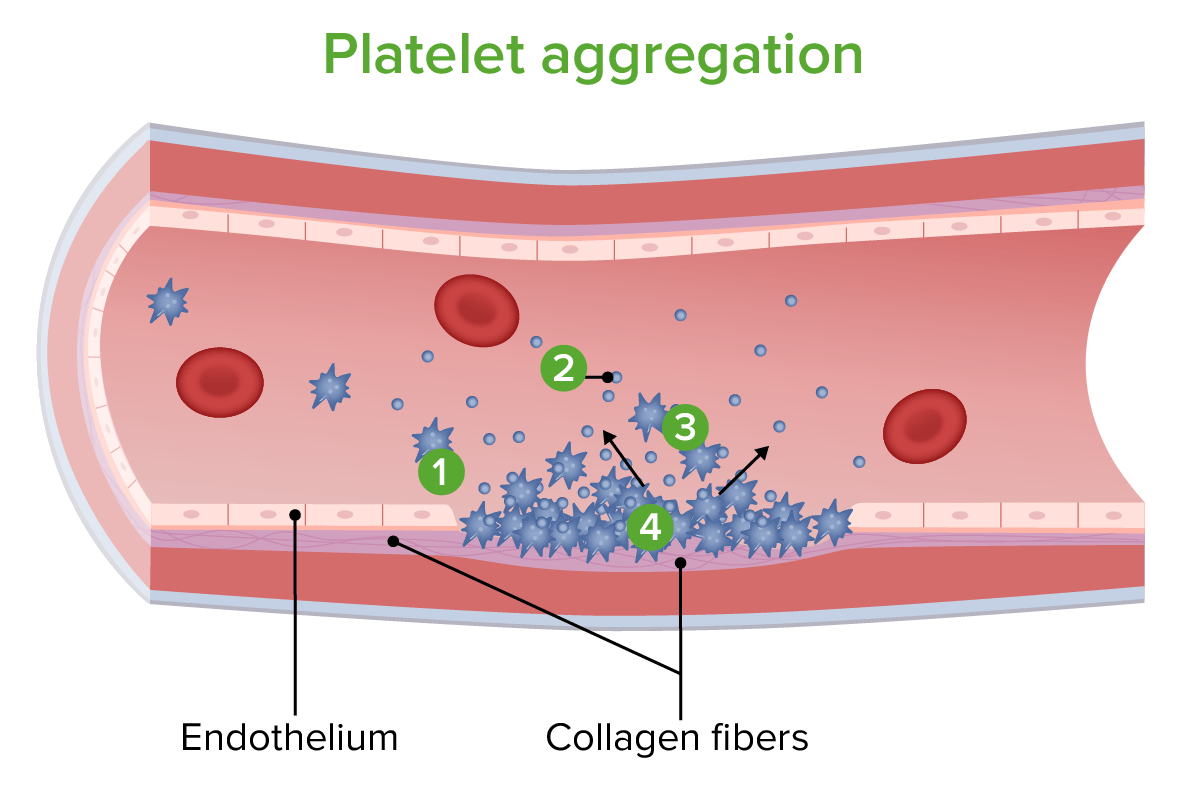
Platelet aggregation:
1. Platelets adhere to exposed collagen fibers.
2. Chemicals are released by platelets to induce vasoconstriction and to attract more platelets.
3. More platelets gather.
4. Platelets cluster to repair the vessel wall.
Image by Lecturio.
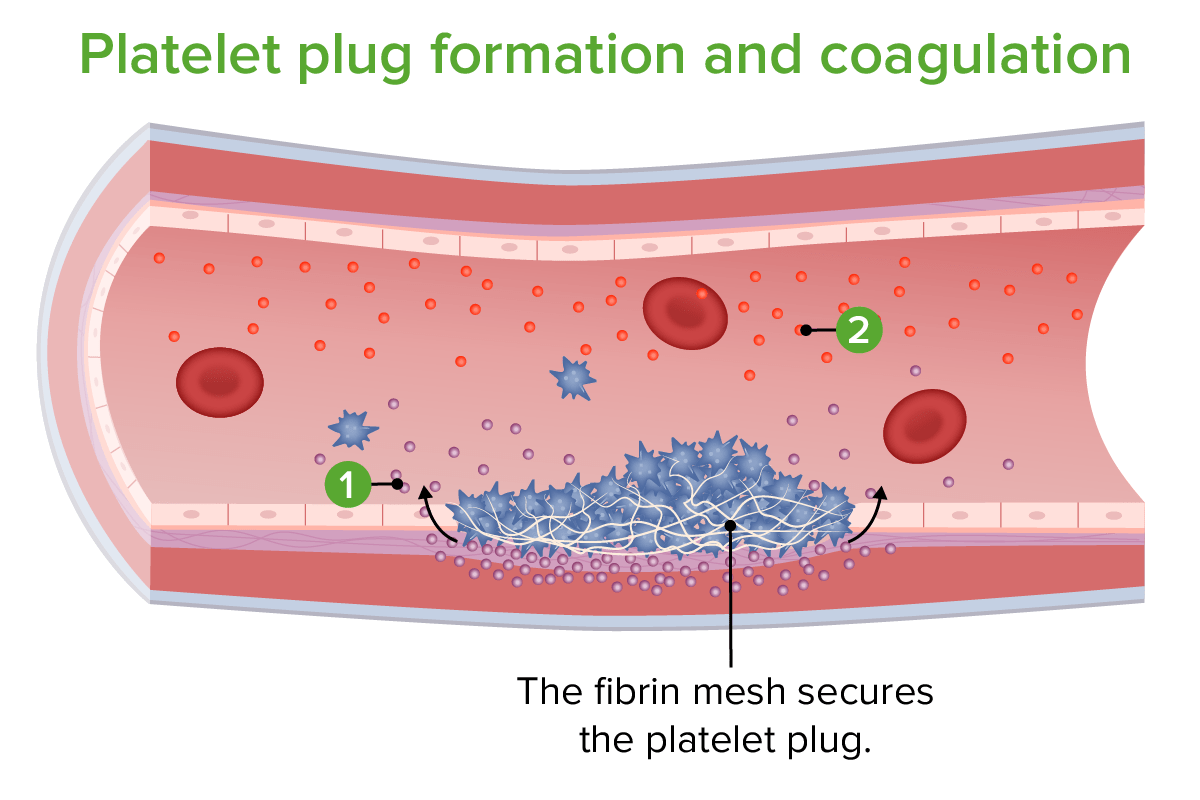
Platelet plug formation and coagulation:
1. Tissue factor released
2. Clotting factors released
Image by Lecturio.
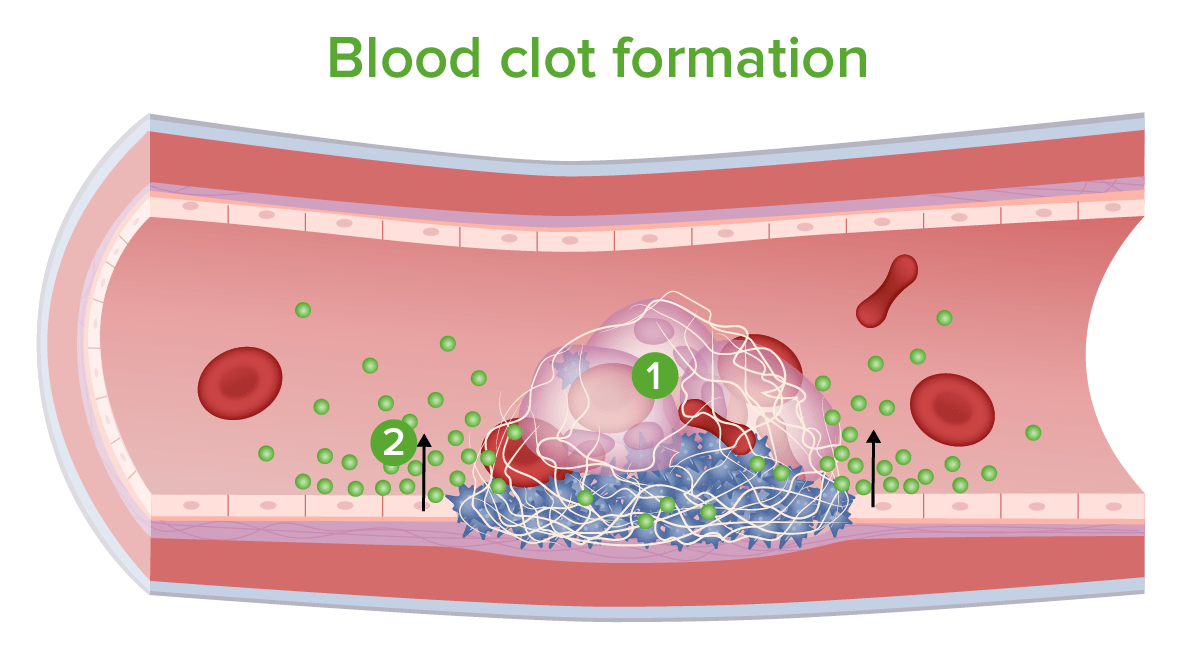
Blood clot formation:
1. Red and white blood cells are trapped in mesh.
2. Coagulation inhibitors and other chemicals are released.
Image by Lecturio.
Vasoconstriction and Formation of the Platelet Plug
Neural stimulation reflex: innate contraction of the vascular smooth musclesSmooth musclesUnstriated and unstriped muscle, one of the muscles of the internal organs, blood vessels, hair follicles, etc. Contractile elements are elongated, usually spindle-shaped cells with centrally located nuclei. Smooth muscle fibers are bound together into sheets or bundles by reticular fibers and frequently elastic nets are also abundant.Muscle Tissue: Histology upon injury
Endothelin: a vasoconstrictorsecreted from the damaged endothelial cells
Thromboxane: a vasoconstrictor released from plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Steps in formation of the platelet plug
Following an endothelial cell injuryCell injuryThe cell undergoes a variety of changes in response to injury, which may or may not lead to cell death. Injurious stimuli trigger the process of cellular adaptation, whereby cells respond to withstand the harmful changes in their environment. Overwhelmed adaptive mechanisms lead to cell injury. Mild stimuli produce reversible injury. If the stimulus is severe or persistent, injury becomes irreversible. Cell Injury and Death, the following processes occur with the plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology to form a temporary platelet plug (also known as primary hemostasis):
AdhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies
Activation
AggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
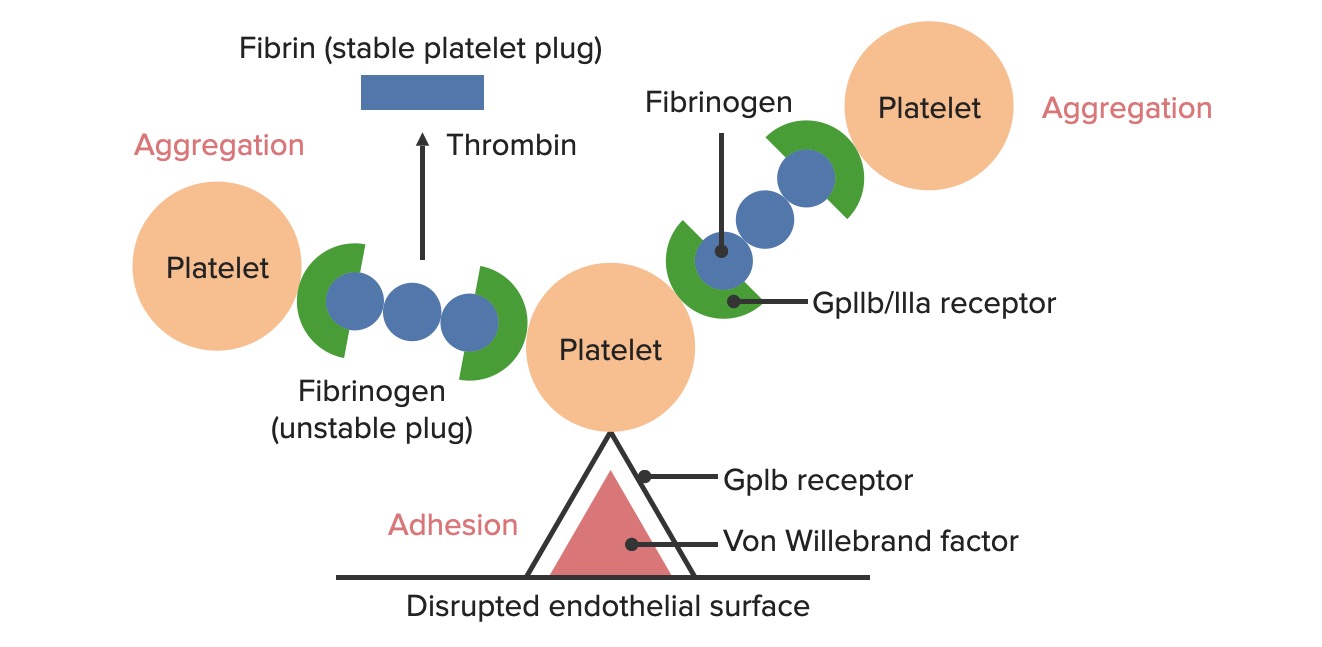
Formation of the temporary hemostatic plug: The disrupted endothelial surface exposes von Willebrand factor (vWF) to the passing blood. Platelets bind to vWF via their GpIb receptors and are activated. Platelet activation triggers the secretion of ADP, which stimulates the expression of the GpIIb/IIIa receptors on the platelets. The GpIIb/IIIa receptors bind to fibrinogen and a platelet on each end, causing platelets to aggregate. As more platelets bind to each other, a platelet plug is formed. As the coagulation cascade is activated, thrombin converts the weaker fibrinogen into the stronger fibrin, creating a much more stable clot.
Image by Lecturio.
Platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies
Exposure of the blood to subendothelialSubendothelialMembranoproliferative Glomerulonephritis components at the site of injury causes plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology to adhere to the injury site.
GpIbreceptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptorson the plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: HistologybindBINDHyperbilirubinemia of the Newborn to the exposed von Willebrand factor (vWF) within the subendothelialSubendothelialMembranoproliferative Glomerulonephritis matrix. This bond is strong enough to withstand the shearing force of the flowing blood.
Other adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies interactions occur:
Involve collagenCollagenA polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth).Connective Tissue: Histology, other glycoprotein receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, and tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids kinase receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors
Contribute to both adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies and activation of plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Adherent plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology are activated.
Platelet activation
Activated plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology enhance further platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies, and stimulate secretionSecretionCoagulation Studies.
Platelet activators:
Potent platelet activators:
Thrombin: produced in the coagulation cascade
CollagenCollagenA polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth).Connective Tissue: Histology: interacts with plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology at the site of injury
Weaker platelet activators:
ADP: acts in an autocrine fashion → released by plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology to help activate other plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
EpinephrineEpinephrineThe active sympathomimetic hormone from the adrenal medulla. It stimulates both the alpha- and beta- adrenergic systems, causes systemic vasoconstriction and gastrointestinal relaxation, stimulates the heart, and dilates bronchi and cerebral vessels.Sympathomimetic Drugs
Activated plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology:
Undergo shape change to become an elongated pseudopod → new shape is extremely adherent
Activate their GpIIb/IIIa receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors so that they are capable of binding to fibrinogen
Release their granules (see “Platelet secretion” below) → assists in activation of the coagulation cascade
Platelet aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
GpIIb/IIIa receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors present on the activated plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology begin binding to fibrinogen.
Fibrinogen is a symmetricalSymmetricalDermatologic Examination molecule that can bindBINDHyperbilirubinemia of the Newborn 2 plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology simultaneously (1 on each end of the fibrinogen).
Forms fibrinogen bridges between plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Results in platelet aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies and formation of a primary hemostatic plug
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology contain 2 types of granules. These granules release various substances when plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology are activated.
Functions of secreted substances:
Recruit and activate additional plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Stimulate expression of GpIIb/IIIa on plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology → enhanced aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
Contribute to initiation of the coagulation cascade
Alpha granules contain:
Fibrinogen
vWF
Factor V (part of the common pathway of the coagulation cascade)
Platelet-derived growth factorPlatelet-derived growth factorMitogenic peptide growth hormone carried in the alpha-granules of platelets. It is released when platelets adhere to traumatized tissues. Connective tissue cells near the traumatized region respond by initiating the process of replication.Hypertrophic and Keloid Scars (PDGF)
Platelet factor-4
FibronectinFibronectinGlycoproteins found on the surfaces of cells, particularly in fibrillar structures. The proteins are lost or reduced when these cells undergo viral or chemical transformation. They are highly susceptible to proteolysis and are substrates for activated blood coagulation factor VIII. The forms present in plasma are called cold-insoluble globulins.Connective Tissue: Histology
Thrombospondin
Dense granules contain:
ADP
SerotoninSerotoninA biochemical messenger and regulator, synthesized from the essential amino acid l-tryptophan. In humans it is found primarily in the central nervous system, gastrointestinal tract, and blood platelets. Serotonin mediates several important physiological functions including neurotransmission, gastrointestinal motility, hemostasis, and cardiovascular integrity.Receptors and Neurotransmitters of the CNS
Histamine
CalciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes
The coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis clot. This process is also known as secondary hemostasis.
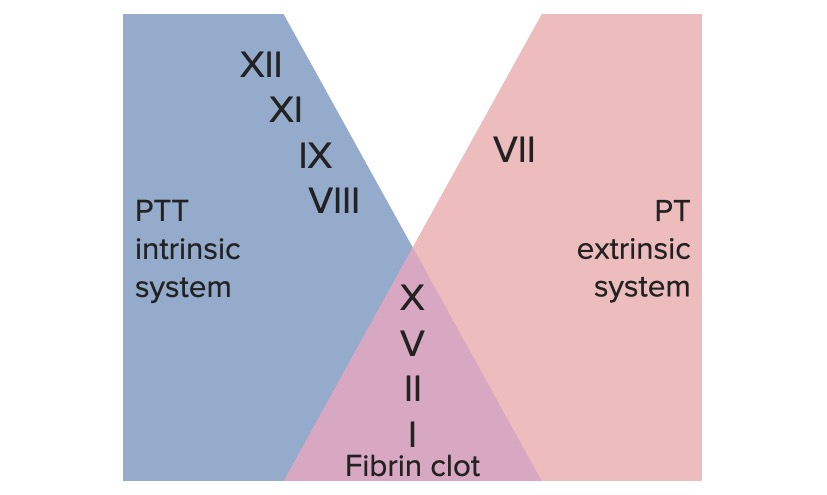
Extrinsic pathway: primarily responsible for initiation of the cascade
Intrinsic pathway: primarily involved in amplification of the cascade
Common pathway:
The extrinsic and intrinsic pathways join together to form the final common pathway when factor X is activated.
Formation of the fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis clot occurs at the end of the common pathway.
Initiation:
The extrinsic pathway is activated with endothelial injury and ultimately produces activated factor X (Xa).
Factor Xa then moves through the common pathway.
Thrombin is produced in the common pathway.
Amplification:
The initial production of thrombin activates multiple factors in the intrinsic and common pathways.
As the intrinsic pathway is activated, an increased amount of factor Xa is produced.
Factor Xa allows for increased activation of the common pathway:
More fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis is produced → propagates the clot
More thrombin is produced → positive feedback loops
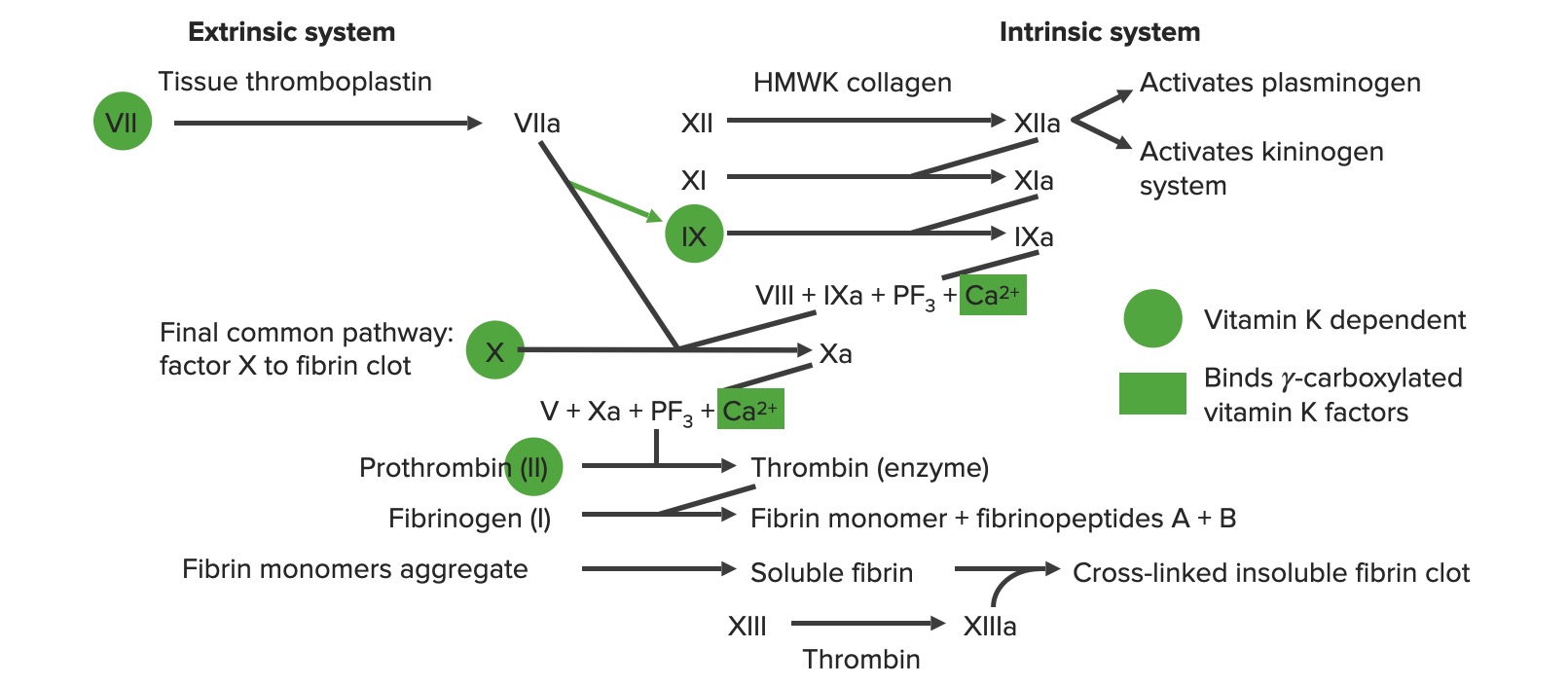
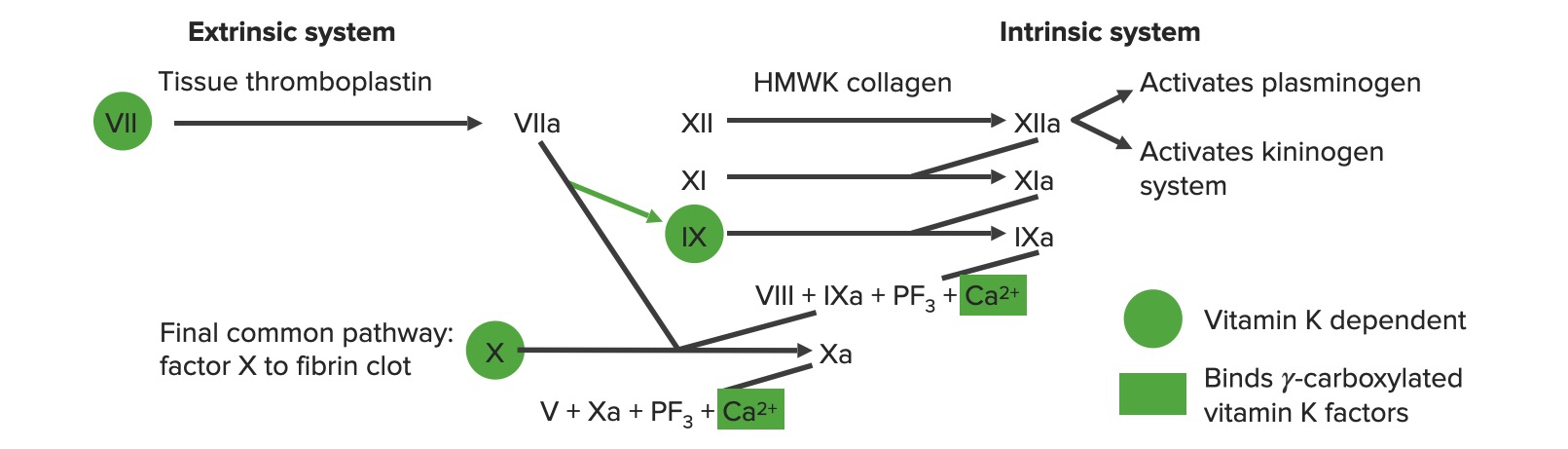
Overview of the coagulation cascade a: activated form PF3: platelet factor 3 (phospholipids)
Image by Lecturio.
Coagulation factors
Coagulation factors are trypsin-like serineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino AcidsproteasesProteasesProteins and Peptides and are denoted with roman numerals.
All procoagulant factors are synthesized in the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy except:
Factor VIII: produced in endothelial cells
vWF: produced in the megakaryocytes and endothelial cells
Vitamin K-dependent factors:
Undergo carboxylation to become functional, require vitamin KVitamin KA lipid cofactor that is required for normal blood clotting. Several forms of vitamin K have been identified: vitamin K 1 (phytomenadione) derived from plants, vitamin K 2 (menaquinone) from bacteria, and synthetic naphthoquinone provitamins, vitamin K 3 (menadione). Vitamin k 3 provitamins, after being alkylated in vivo, exhibit the antifibrinolytic activity of vitamin k. Green leafy vegetables, liver, cheese, butter, and egg yolk are good sources of vitamin k.Fat-soluble Vitamins and their Deficiencies
Procoagulants:
Factor II
Factor VII
Factor IX
Factor X
AnticoagulantsAnticoagulantsAnticoagulants are drugs that retard or interrupt the coagulation cascade. The primary classes of available anticoagulants include heparins, vitamin K-dependent antagonists (e.g., warfarin), direct thrombin inhibitors, and factor Xa inhibitors. Anticoagulants:
Protein C
Protein S
Vitamin KVitamin KA lipid cofactor that is required for normal blood clotting. Several forms of vitamin K have been identified: vitamin K 1 (phytomenadione) derived from plants, vitamin K 2 (menaquinone) from bacteria, and synthetic naphthoquinone provitamins, vitamin K 3 (menadione). Vitamin k 3 provitamins, after being alkylated in vivo, exhibit the antifibrinolytic activity of vitamin k. Green leafy vegetables, liver, cheese, butter, and egg yolk are good sources of vitamin k.Fat-soluble Vitamins and their Deficiencies:
Primarily synthesized in the colonColonThe large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy
Activated by epoxide reductase in the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy
Works as a cofactor for gamma-glutamyl carboxylase to carboxylate the Vitamin-K dependent factors
These carboxylated factors gain affinity for the negatively charged phospholipidsPhospholipidsLipids containing one or more phosphate groups, particularly those derived from either glycerol (phosphoglycerides) or sphingosine (sphingolipids). They are polar lipids that are of great importance for the structure and function of cell membranes and are the most abundant of membrane lipids, although not stored in large amounts in the system.Lipid Metabolism on plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology → promote coagulation
Form multicomponent enzyme complexes which:
Perform critical steps in the coagulation cascade
Each contain a proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS, a cofactor, and a substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes
Bound to anionic phospholipid membrane surfaces
Restrict a majority of thrombin generation to the sites of vascular injury
Factor VIIa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + tissue factor (cofactor) + factor X (substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes)
Activates factor X → factor Xa
Intrinsic X-ase:
Factor IXa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + factor VIIIa (cofactor) + factor X (substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes)
Activates factor X → factor Xa
Prothrombinase:
Factor Xa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + factor VaVAVentilation: Mechanics of Breathing (cofactor) + prothrombin (substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes)
Activates prothrombin → thrombin
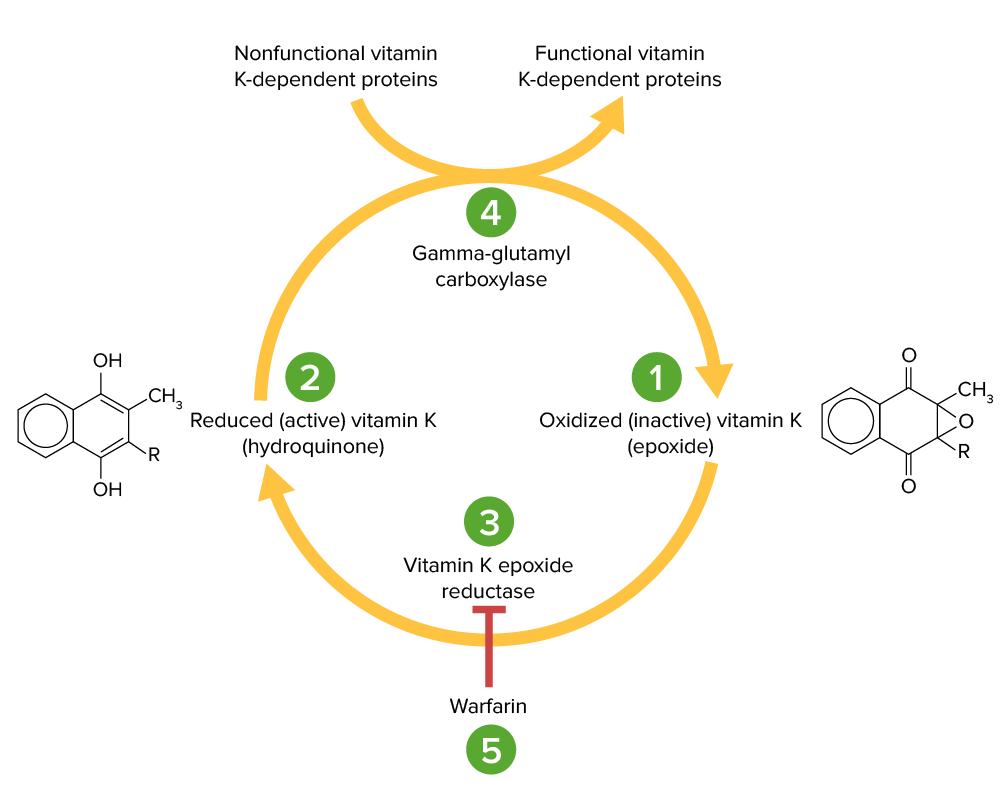
Vitamin K cycle:
Vitamin K epoxide (1) is inactive and converted to its active, reduced form, vitamin K hydroquinone (2), by vitamin K epoxide reductase (VKOR; 3). Vitamin K hydroquinone is a cofactor in the carboxylation of specific glutamate residues within the vitamin K-dependent proteins (factors II, VII, IX, X, protein C and S), a process which is necessary to activate them. The carboxylation reaction is catalyzed by gamma-glutamyl carboxylase (4). Vitamin K hydroquinone is oxidized to the epoxide form when it acts as a cofactor, but is then recycled back to the hydroquinone form by VKOR. Warfarin inhibits VKOR (5) so that vitamin K cannot be recycled from its oxidized form to the reduced form. Thus, vitamin K-dependent proteins cannot be activated.
Image by Lecturio.
Extrinsic pathway: The tissue factor pathway
The extrinsic pathway is the primary physiological mechanism by which clotting is initiated.
Factor VIIa activates factor X → Xa. Factor Xa is the 1st step in the common pathway.
To summarize, tissue factor activates VII → VIIa, which activates X → Xa → common pathway
Intrinsic pathway: The contact pathway
The intrinsic pathway is mainly responsible for the amplification of factor X activation. Factor X is activated by the initial thrombin generated by the extrinsic/common pathway, but also can be activated directly by endothelial injury.
Exposure to negatively charged collagenCollagenA polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth).Connective Tissue: Histology in the subendothelialSubendothelialMembranoproliferative Glomerulonephritis matrix activates high-molecular-weight kininogen (HMWK) and prekallikrein (PK).
HMWK + PK activate factor XII → XIIa
Factor XIIaFactor XIIaActivated form of factor XII. In the initial event in the intrinsic pathway of blood coagulation, kallikrein (with cofactor high molecular weight kininogen) cleaves factor XII to XIIa. Factor XIIa is then further cleaved by kallikrein, plasmin, and trypsin to yield smaller factor XII fragments (hageman-factor fragments). These fragments increase the activity of prekallikrein to kallikrein but decrease the procoagulant activity of factor XII.Inflammation activates:
Factor XI → XIa
Thrombin (from the common pathway) also activates factor XI.
Prekallikrein → kallikreinKallikreinProteolytic enzymes from the serine endopeptidase family found in normal blood and urine. Specifically, kallikreins are potent vasodilators and hypotensives and increase vascular permeability and affect smooth muscle. They act as infertility agents in men. Three forms are recognized, plasma kallikrein, tissue kallikrein, and prostate-specific antigen.Hereditary Angioedema (C1 Esterase Inhibitor Deficiency)
KallikreinKallikreinProteolytic enzymes from the serine endopeptidase family found in normal blood and urine. Specifically, kallikreins are potent vasodilators and hypotensives and increase vascular permeability and affect smooth muscle. They act as infertility agents in men. Three forms are recognized, plasma kallikrein, tissue kallikrein, and prostate-specific antigen.Hereditary Angioedema (C1 Esterase Inhibitor Deficiency) augments further activation of XII → XIIa
Factor XIa activates factor IX → IXa
Intrinsic X-ase: factor IXa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) combines with factor VIIIa (cofactor) to activate factor X (substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes) → Xa
Factor VIII:
Activated by factor Xa and thrombin (both initially generated by the extrinsic and common pathways)
Stabilized by vWF
Factor Xa is the 1st step in the common pathway.
To summarize, HMWK + PK activate → 12, which activates → 11, which activates → 9, which combines with 8 to activate → 10
The extrinsic and intrinsic coagulation systems
Image by Lecturio.
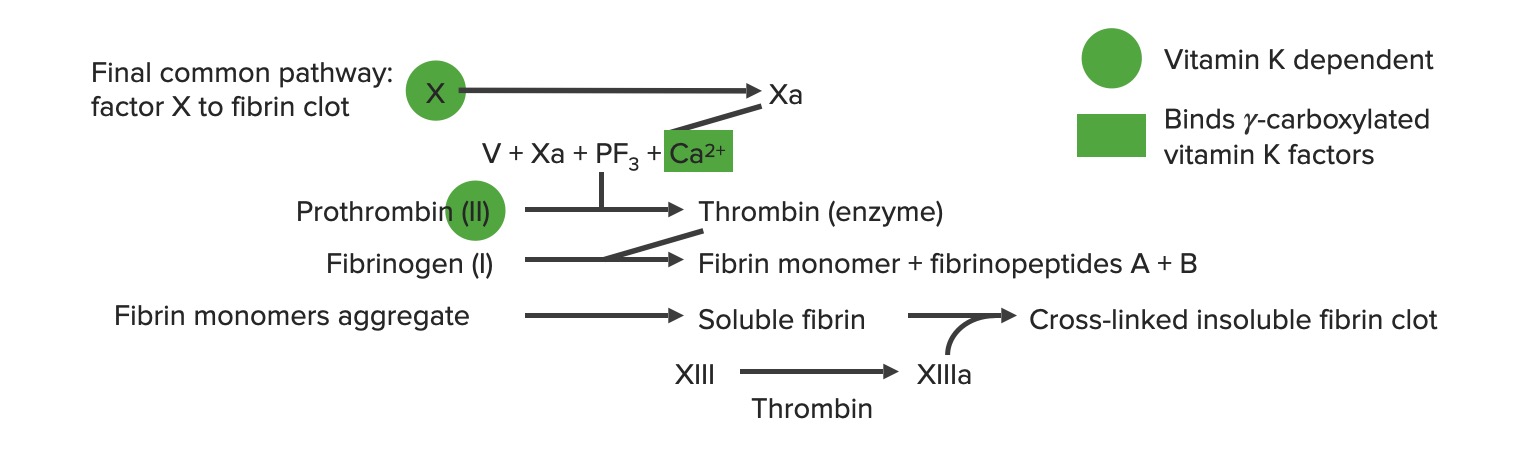
Common pathway
Begins with prothrombinase: factor Xa combines with factor VaVAVentilation: Mechanics of Breathing and calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes to activate prothrombin (factor II) → thrombin (factor IIa)
Thrombin (factor IIa) activates the following:
Fibrinogen (factor I) → fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis (factor Ia) → clot propagationPropagationPropagation refers to how the electrical signal spreads to every myocyte in the heart.Cardiac Physiology
Factor XIII → XIIIa → cross-linking of fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis polymers to stabilize the clot
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology → activated plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology → aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies and secretionSecretionCoagulation Studies
Generation of thrombin leads to multiple positive feedback loops → ↑↑ production of thrombin
The final common pathway a: activated form PF3: platelet factor 3 (phospholipids)
The body produces several substances that inhibit platelet binding, aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies, and secretionSecretionCoagulation Studies, as well as function as natural anticoagulantsAnticoagulantsAnticoagulants are drugs that retard or interrupt the coagulation cascade. The primary classes of available anticoagulants include heparins, vitamin K-dependent antagonists (e.g., warfarin), direct thrombin inhibitors, and factor Xa inhibitors. Anticoagulants. These mechanisms limitLimitA value (e.g., pressure or time) that should not be exceeded and which is specified by the operator to protect the lungInvasive Mechanical Ventilation clotting to specific focal sites and keep the blood fluid.
Tissue factor pathway inhibitor (TFPI):
Inhibits the activation of factor X
Located primarily on the surface of microvascular endothelial cells
AntithrombinAntithrombinEndogenous factors and drugs that directly inhibit the action of thrombin, usually by blocking its enzymatic activity. They are distinguished from indirect thrombin inhibitors, such as heparin, which act by enhancing the inhibitory effects of antithrombins.Anticoagulants:
Natural circulating anticoagulant produced by the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy
Inhibits activated forms of factors II, IX, and X
Rate of factor inactivation is augmented by heparin
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis C and S:
Vitamin K-dependent factors produced by the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy
Protein C cleaves and inactivates factors V and VIII.
Protein S augments the activity of protein C.
Other anticoagulant substances produced by endothelial cells:
ProstacyclinProstacyclinA prostaglandin that is a powerful vasodilator and inhibits platelet aggregation. It is biosynthesized enzymatically from prostaglandin endoperoxides in human vascular tissue. The sodium salt has been also used to treat primary pulmonary hypertension.Eicosanoids: a vasodilator that blocks platelet aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
Nitric oxideNitric OxideA free radical gas produced endogenously by a variety of mammalian cells, synthesized from arginine by nitric oxide synthase. Nitric oxide is one of the endothelium-dependent relaxing factors released by the vascular endothelium and mediates vasodilation. It also inhibits platelet aggregation, induces disaggregation of aggregated platelets, and inhibits platelet adhesion to the vascular endothelium. Nitric oxide activates cytosolic guanylate cyclase and thus elevates intracellular levels of cyclic gmp.Pulmonary Hypertension Drugs: a vasodilator that blocks platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
Thrombomodulin: binds to thrombin and converts it into an anticoagulant that activates protein C
The fibrinolytic system functions to remove the clot after the vasculature is repaired, and the process is accomplished primarily by plasmin.
Plasmin: cleaves fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis polymers (fibrinolysis)
Plasminogen is activated (converted to plasmin) by:
Urine plasminogen activator (UPa) aka urokinaseUrokinaseA proteolytic enzyme that converts plasminogen to fibrinolysin where the preferential cleavage is between arginine and valine. It was isolated originally from human urine, but is found in most tissues of most vertebrates.Thrombolytics
Generates new plasmin-binding sites on partially degraded fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis
Time taken for the plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot when exposed to tissue factor
Measures function of the extrinsic and common pathways
Normal range: approximately 11–13 seconds
Elevated in:
WarfarinWarfarinAn anticoagulant that acts by inhibiting the synthesis of vitamin K-dependent coagulation factors. Warfarin is indicated for the prophylaxis and/or treatment of venous thrombosis and its extension, pulmonary embolism, and atrial fibrillation with embolization. It is also used as an adjunct in the prophylaxis of systemic embolism after myocardial infarction. Warfarin is also used as a rodenticide.Anticoagulants therapy
Vitamin K deficiencyVitamin K DeficiencyA nutritional condition produced by a deficiency of vitamin K in the diet, characterized by an increased tendency to hemorrhage (hemorrhagic disorders). Such bleeding episodes may be particularly severe in newborn infants.Fat-soluble Vitamins and their Deficiencies
Deficiency of factors II, V, VII, and X
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease
Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation (DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation)
INR:
A ratio comparing the patient’s PT to a reference PT
Measures function of the extrinsic and common pathways
Normal range: approximately 0.8–1.1
PTT:
Time taken for the plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot when exposed to a negatively charged substance (which activates the intrinsic pathway)
Measures function of both the intrinsic and common pathways
Normal range: 25–40 sec
Elevated in:
Heparin therapy
HemophiliaHemophiliaThe hemophilias are a group of inherited, or sometimes acquired, disorders of secondary hemostasis due to deficiency of specific clotting factors. Hemophilia A is a deficiency of factor VIII, hemophilia B a deficiency of factor IX, and hemophilia C a deficiency of factor XI. Patients present with bleeding events that may be spontaneous or associated with minor or major trauma.Hemophilia (abnormal factor VIII or IX)
von Willebrand diseaseVon Willebrand diseaseVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease (vWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease)
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
Bleeding time (BT):
Measures platelet function
Normal range: 2–7 minutes
Prolonged in:
ThrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
vWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease
Bernard-Soulier disease
Glanzmann’s thrombasthenia
Renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
NSAIDNSAIDNonsteroidal antiinflammatory drugs (NSAIDs) are a class of medications consisting of aspirin, reversible NSAIDs, and selective NSAIDs. NSAIDs are used as antiplatelet, analgesic, antipyretic, and antiinflammatory agents. Nonsteroidal Antiinflammatory Drugs (NSAIDs) and/or aspirinAspirinThe prototypical analgesic used in the treatment of mild to moderate pain. It has anti-inflammatory and antipyretic properties and acts as an inhibitor of cyclooxygenase which results in the inhibition of the biosynthesis of prostaglandins. Aspirin also inhibits platelet aggregation and is used in the prevention of arterial and venous thrombosis.Nonsteroidal Antiinflammatory Drugs (NSAIDs) use
Released upon cleavage of cross-linked fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis by plasmin
Indicates recent or ongoing coagulation and fibrinolysis
Disorders of primary hemostasis (formation of the platelet plug)
Glanzmann thrombastheniaGlanzmann ThrombastheniaHypocoagulable Conditions: an autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance bleeding syndrome characterized by a deficiency of the GpIIb/IIIa receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, resulting in a lack of platelet aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
Bernard-Soulier syndrome: an autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance bleeding syndrome characterized by deficiency of the GpIb receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, resulting in failure of platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies. Bernard-Soulier syndrome can be diagnosed using a ristocetin assay. Ristocetin activates vWF to allow binding to the platelet GpIb receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors; however, in Bernard-Soulier syndrome, the plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology will fail to adhere in the assay.
Immune thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia: an autoimmune disorderAutoimmune DisorderSeptic Arthritis characterized by anti-GpIIb/IIIa auto-antibodies, which lead to the destruction of plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology. Immune thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia often occurs after GI or respiratory viral infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, although it can also be a drug-induced condition. Clinically, immune thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia may present with prolonged bleeding, petechiaePetechiaePrimary Skin Lesions, easy bruisingEasy bruisingChédiak-Higashi Syndrome, and/or purpura. Treatment may include a platelet transfusion or splenectomySplenectomySurgical procedure involving either partial or entire removal of the spleen.Rupture of the Spleen, or management with steroidsSteroidsA group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus.Benign Liver Tumors and IV immunoglobulinsIV immunoglobulinsImmunoglobulin preparations used in intravenous infusion, containing primarily immunoglobulin g. They are used to treat a variety of diseases associated with decreased or abnormal immunoglobulin levels including pediatric aids; primary hypergammaglobulinemia; scid; cytomegalovirus infections in transplant recipients, lymphocytic leukemia, chronic; kawasaki syndrome, infection in neonates, and idiopathic thrombocytopenic purpura.DiGeorge Syndrome.
Thrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of ADAMTS-13, a metalloproteinase that cleaves multimers of von Willebrand factor (VWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets. Thrombotic Thrombocytopenic Purpura (TTPTTPThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of adamts-13, a metalloproteinase that cleaves multimers of von Willebrand factor (vWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets.Thrombotic Thrombocytopenic Purpura): a bleeding disorder marked by a pentad of feverFeverFever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever, microangiopathic hemolytic anemiaMicroangiopathic Hemolytic AnemiaHemolytic Uremic Syndrome, thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome, and neurological symptoms. Thrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of ADAMTS-13, a metalloproteinase that cleaves multimers of von Willebrand factor (VWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets. Thrombotic Thrombocytopenic Purpura occurs due to a congenitalCongenitalChorioretinitis or acquired deficiency of ADAMTS-13, which is a metalloprotease that cleaves the vWF. A deficiency of ADAMTS-13 results in large vWF multimers that increase platelet aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies, leading to microvascular thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus and a consumption of plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology.
Disorders of secondary hemostasis (the coagulation cascade)
HemophiliaHemophiliaThe hemophilias are a group of inherited, or sometimes acquired, disorders of secondary hemostasis due to deficiency of specific clotting factors. Hemophilia A is a deficiency of factor VIII, hemophilia B a deficiency of factor IX, and hemophilia C a deficiency of factor XI. Patients present with bleeding events that may be spontaneous or associated with minor or major trauma.Hemophilia: a rare blood-clotting disorder in which the body lacks blood-clotting factors (factor VIII in hemophilia AHemophilia AThe classic hemophilia resulting from a deficiency of factor VIII. It is an inherited disorder of blood coagulation characterized by a permanent tendency to hemorrhage.Hemophilia; factor IX in hemophilia BHemophilia BA deficiency of blood coagulation factor IX inherited as an X-linked disorder. (also known as Christmas disease, after the first patient studied in detail, not the holiday.) historical and clinical features resemble those in classic hemophilia, but patients present with fewer symptoms. Severity of bleeding is usually similar in members of a single family. Many patients are asymptomatic until the hemostatic system is stressed by surgery or trauma. Treatment is similar to that for hemophilia A.Hemophilia). Affected individuals present with abnormal bleeding that can occur spontaneously or after minor trauma. These individuals can bleed into joint spaces and develop life-threatening internal bleeding.
Mixed disorders affecting both plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology and coagulation factors
vWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease: the most commonly inherited disorder of hemostasis caused by a qualitative or quantitative deficiency of von Willebrand factor. There are 3 primary types, which differ in severity, although all tend to present with bleeding abnormalities. Von Willebrand factor is required for both initial platelet adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies and to help stabilize factor VIII in the intrinsic pathway.
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation: a serious medical condition in which the coagulation cascade is activated systemically, leading to multiple clots that can lead to permanent end-organ damage. During DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation, the coagulation factors are completely consumed. Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation always has a secondary cause. InfectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, burnsBurnsA burn is a type of injury to the skin and deeper tissues caused by exposure to heat, electricity, chemicals, friction, or radiation. Burns are classified according to their depth as superficial (1st-degree), partial-thickness (2nd-degree), full-thickness (3rd-degree), and 4th-degree burns. Burns, and malignancies are among the most common causes. Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation can also occur during a severe postpartum hemorrhagePostpartum hemorrhagePostpartum hemorrhage is one of the most common and deadly obstetric complications. Since 2017, postpartum hemorrhage has been defined as blood loss greater than 1,000 mL for both cesarean and vaginal deliveries, or excessive blood loss with signs of hemodynamic instability. Postpartum Hemorrhage. Laboratory findings include thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, prolongation of PT and PTT, and elevation of D-dimerD-dimerDeep Vein Thrombosis levels.
CirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis: The liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy is the primary site of synthesisSynthesisPolymerase Chain Reaction (PCR) for a majority of the clotting factors. In addition to the impaired synthesisSynthesisPolymerase Chain Reaction (PCR) of clotting factors, cirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis may also independently result in thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia due to splenic sequestrationSplenic sequestrationSevere Congenital Neutropenia of plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology and decreased thrombopoietinThrombopoietinA humoral factor that stimulates the production of thrombocytes (blood platelets). Thrombopoietin stimulates the proliferation of bone marrow megakaryocytes and their release of blood platelets. The process is called thrombopoiesis.Platelets: Histology production. PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology themselves may also be dysfunctional.
Disorders of fibrinolysis
Factor V LeidenFactor V LeidenHypercoagulable StatesmutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations: results in the production of mutant factor V, which is resistant to degradation by activated protein C, thereby leading to increased thrombin production and a procoagulant state in the blood. Complications include deep vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus, cerebral vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus, and pulmonary embolismPulmonary EmbolismPulmonary embolism (PE) is a potentially fatal condition that occurs as a result of intraluminal obstruction of the main pulmonary artery or its branches. The causative factors include thrombi, air, amniotic fluid, and fat. In PE, gas exchange is impaired due to the decreased return of deoxygenated blood to the lungs. Pulmonary Embolism.
Prothrombin gene mutationGene MutationMyotonic Dystrophies: the 2nd most common inherited thrombophiliaInherited ThrombophiliaHypercoagulable States after factor V LeidenFactor V LeidenHypercoagulable States. Point mutations in the prothrombin geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics lead to increased levels of prothrombin, leading to a hypercoagulableHypercoagulableHypercoagulable states (also referred to as thrombophilias) are a group of hematologic diseases defined by an increased risk of clot formation (i.e., thrombosis) due to either an increase in procoagulants, a decrease in anticoagulants, or a decrease in fibrinolysis. Hypercoagulable States state and increased risk of venous thromboembolismThromboembolismObstruction of a blood vessel (embolism) by a blood clot (thrombus) in the blood stream.Systemic Lupus Erythematosus.
Antithrombin deficiencyAntithrombin deficiencyAn absence or reduced level of antithrombin III leading to an increased risk for thrombosis.Budd-Chiari Syndrome: an inherited or acquired disorder resulting in antithrombinAntithrombinEndogenous factors and drugs that directly inhibit the action of thrombin, usually by blocking its enzymatic activity. They are distinguished from indirect thrombin inhibitors, such as heparin, which act by enhancing the inhibitory effects of antithrombins.Anticoagulants activity that is < 80% of its normal activity. Antithrombin deficiencyAntithrombin deficiencyAn absence or reduced level of antithrombin III leading to an increased risk for thrombosis.Budd-Chiari Syndrome leads to a decreased inhibition of factors II, IX, and X, thus, creating a hypercoagulableHypercoagulableHypercoagulable states (also referred to as thrombophilias) are a group of hematologic diseases defined by an increased risk of clot formation (i.e., thrombosis) due to either an increase in procoagulants, a decrease in anticoagulants, or a decrease in fibrinolysis. Hypercoagulable States state.