A hemostase refere-se aos processos corporais inatos e faseados, que ocorrem após a lesão de um vaso, resultando na formação de coágulos e culminando na cessação da hemorragia. A hemostase ocorre em 2 fases, a saber, primária e secundária. A hemostase primária envolve a adesão, ativação e agregação plaquetária ao endotélio vascular danificado, formando um tampão que interrompe a hemorragia, temporariamente. A hemostase secundária envolve a ativação da cascata de coagulação, resultando na formação de um tampão maisMAISAndrogen Insensitivity Syndrome estável. Finalmente, à medida que a vasculatura é reparada, o coágulo é degradado na fase fibrinolítica.
Fase fibrinolítica: para quebrar o coágulo assim que já não for necessário
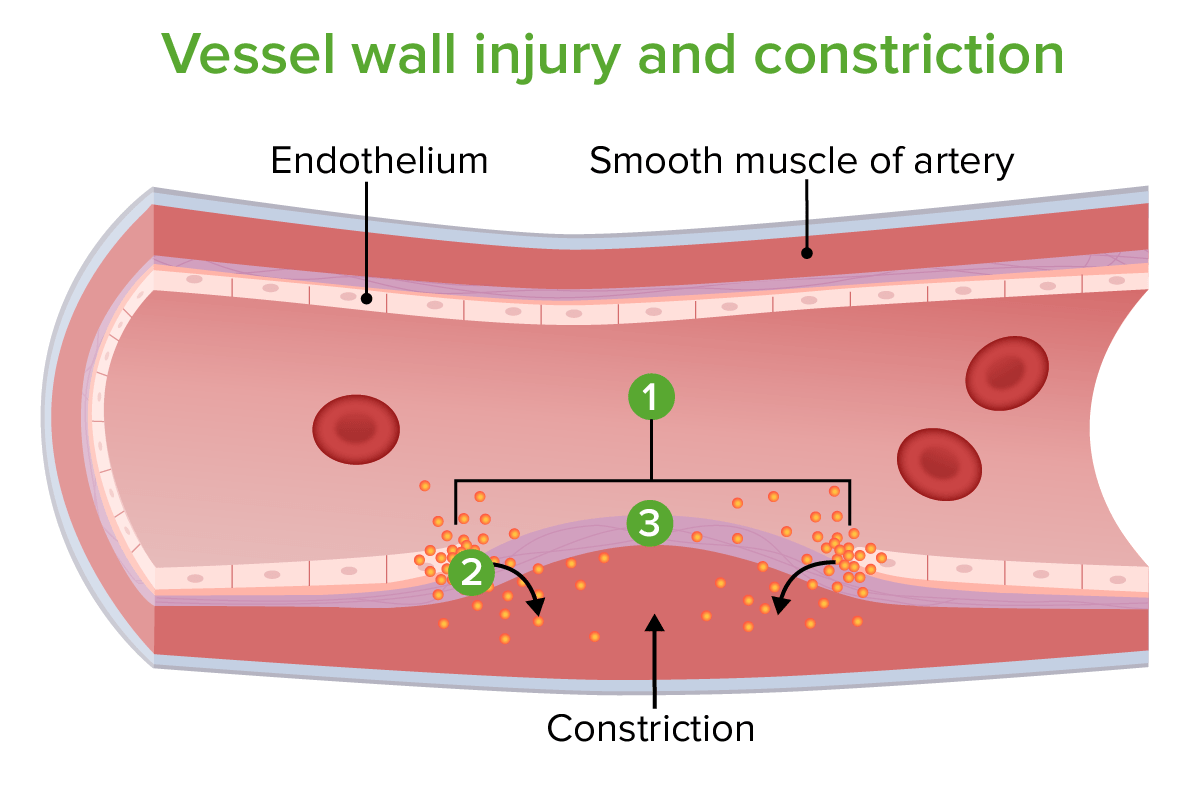
Dano e constrição da parede vascular: 1. Local da lesão 2. Constrição causada por libertação de endotelina 3. Exposição de fibras de colagénio
Imagem por Lecturio.
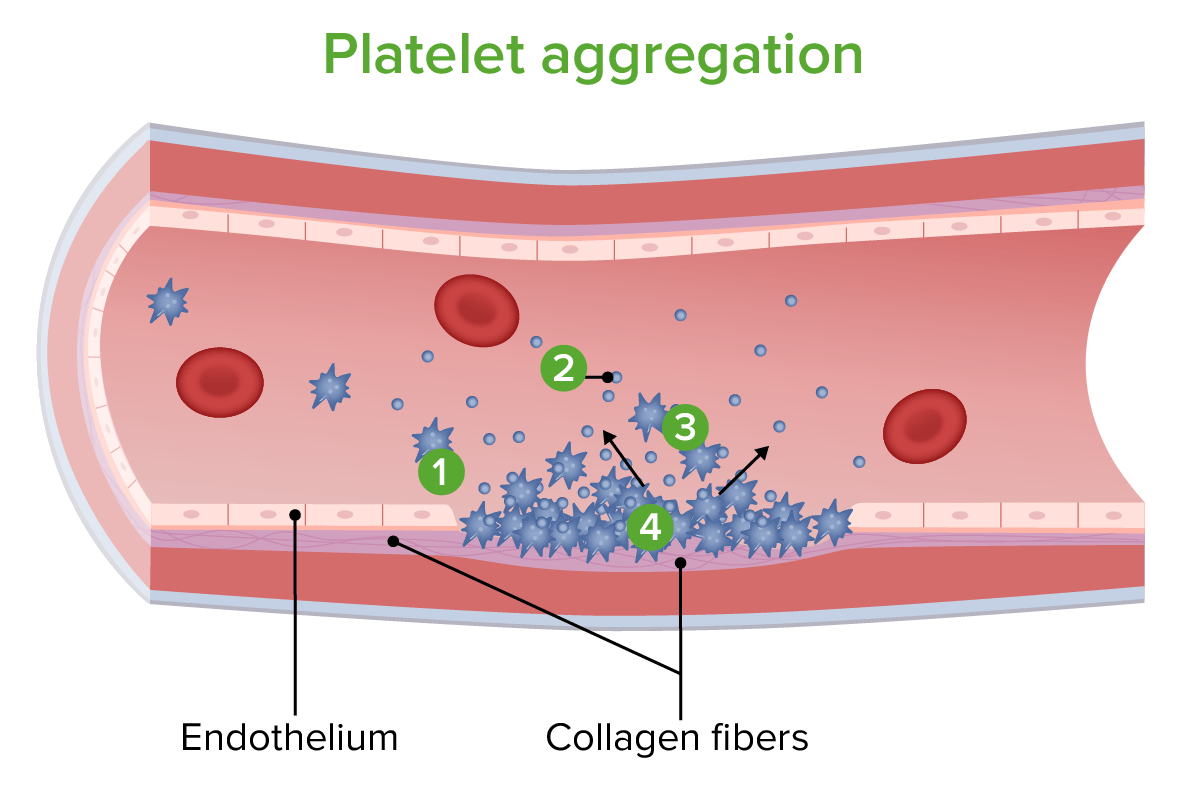
Agregação plaquetária: 1. As plaquetas aderem às fibras de colagénio expostas. 2. As plaquetas libertam químicos para induzir vasoconstrição e para atrair mais plaquetas. 3. Juntam-se mais plaquetas. 4. As plaquetas agregam-se para reparar a parede vascular.
Imagem por Lecturio.
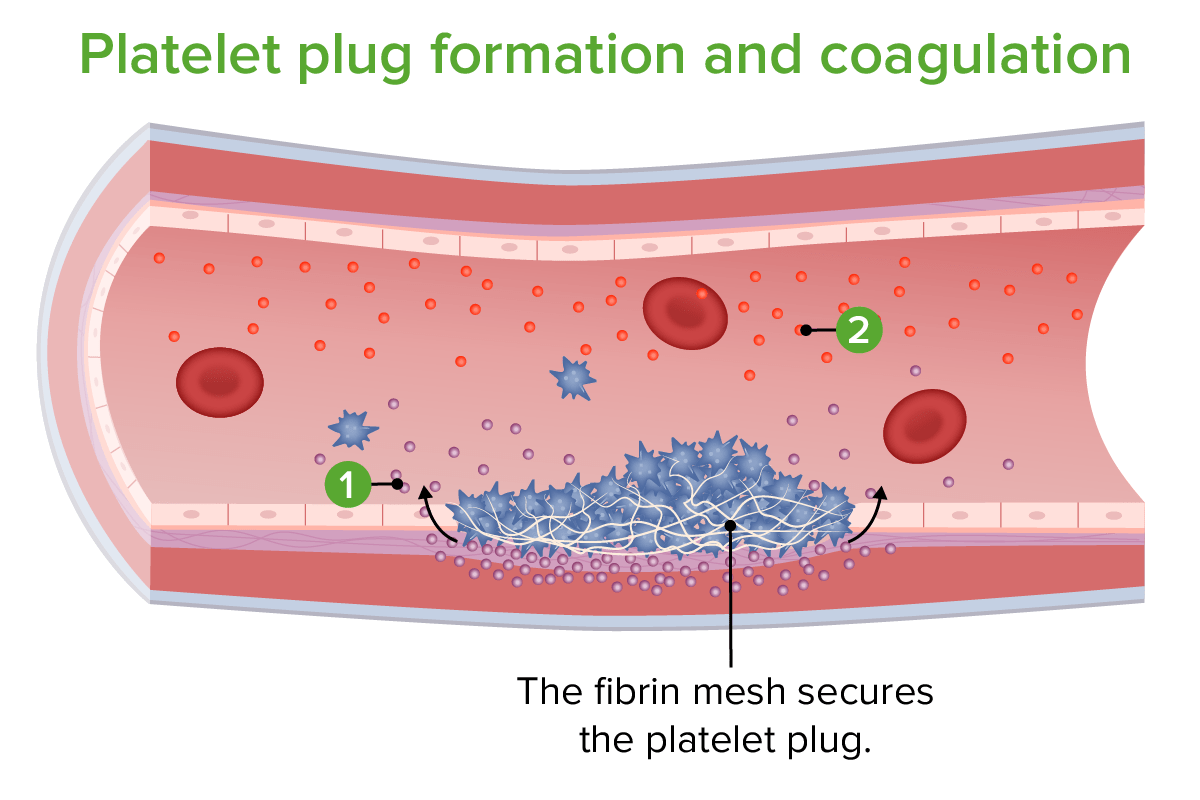
Formação do tampão plaquetário e coagulação: 1. Libertação de fator tecidual 2. Libertação de fatores de coagulação
Imagem por Lecturio.
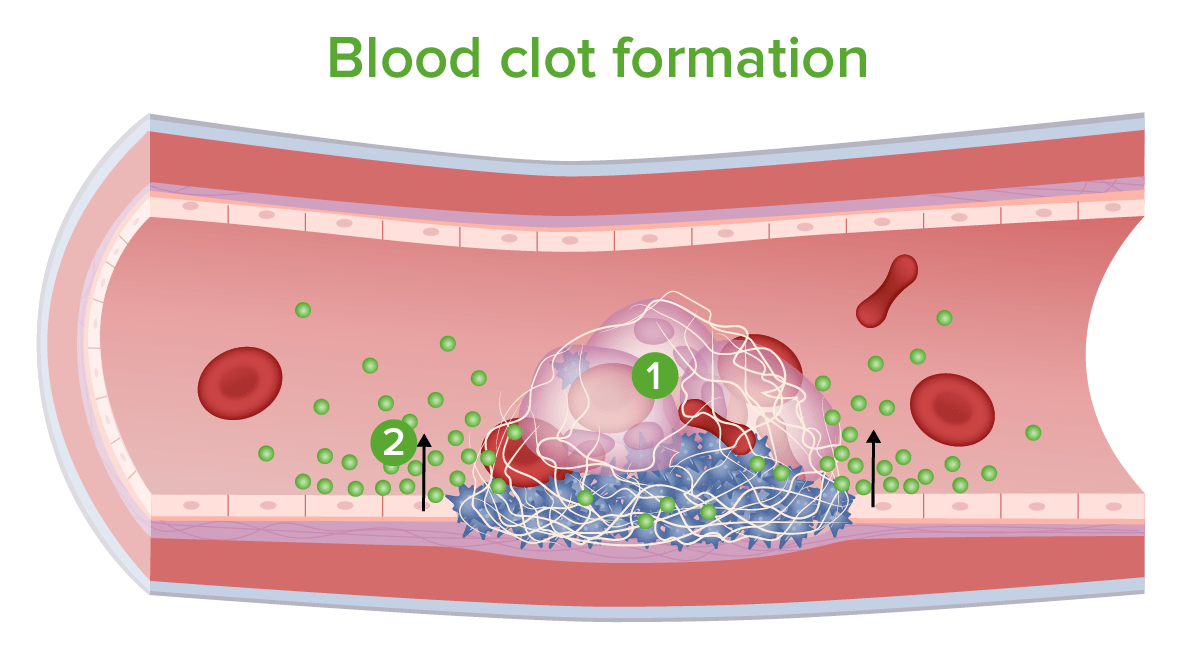
Formação do coágulo sanguíneo: 1. Eritrócitos e leucócitos ficam “presos” na malha. 2. São libertados inibidores da coagulação e outros químicos.
Imagem por Lecturio.
Vasoconstrição e Formação do Tampão Plaquetário
Após uma lesão endotelial, os vasos lesados realizam vasoconstrição. Além disso, a exposição do sangue aos componentes subendoteliais desencadeia a formação do tampão plaquetário.
Vasoconstrição
A lesão endotelial resulta em vasoconstrição transitória via:
Reflexo de estimulação neural: contração inata dos músculos lisos vasculares após a lesão
Endotelina: vasoconstritorsecretado pelas células endoteliais danificadas
Tromboxano: vasoconstritor libertado pelas plaquetas
Etapas na formação do tampão plaquetário
Após uma lesão da célula endotelial, ocorrem os seguintes processos, com as plaquetas a formarem um tampão plaquetário temporário (também conhecido como hemostase primária):
Adesão
Ativação
Agregação
Secreção
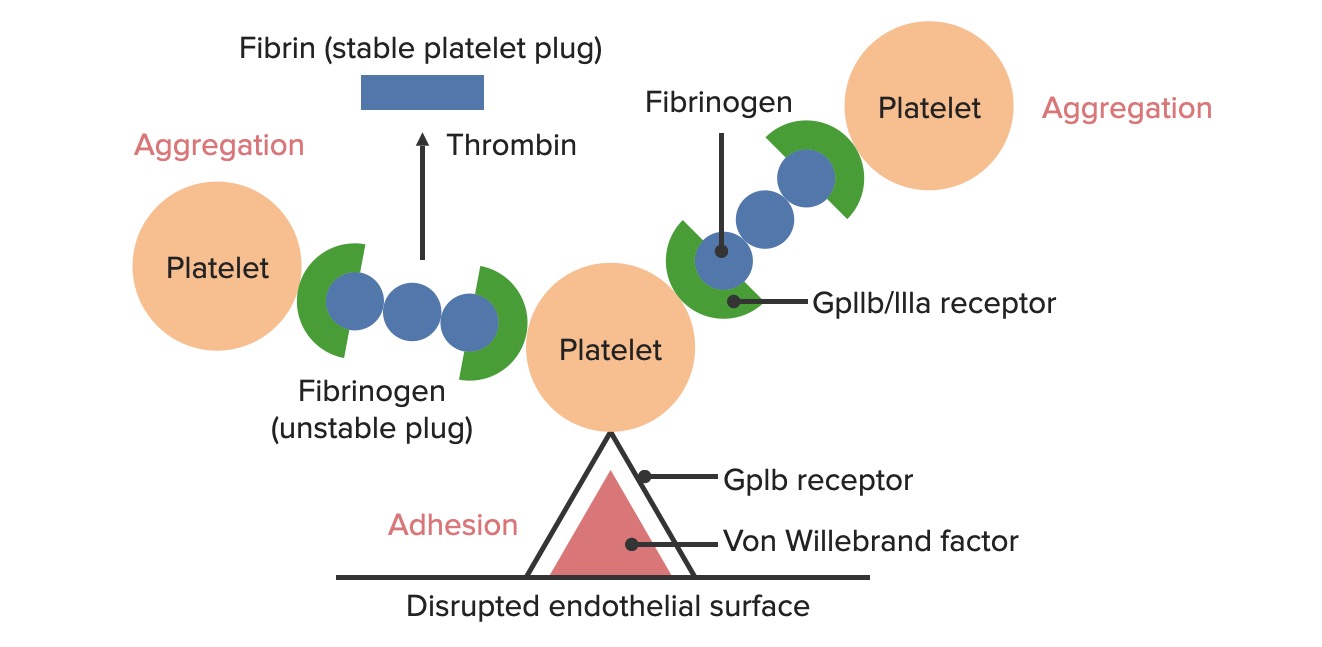
Formação do tampão hemostático temporário: A disrupção da superfície endotelial expõe o fator de von Willebrand (FvW) ao sangue que passa. As plaquetas ligam-se ao FvW, através dos seus recetores GpIb, e são ativadas. A ativação plaquetária desencadeia a secreção de ADP, que estimula a expressão dos recetores GpIIb/IIIa nas plaquetas. Os recetores GpIIb/IIIa ligam-se ao fibrinogénio e a uma plaqueta em cada extremidade, fazendo com que as plaquetas se agreguem. À medida que mais plaquetas se ligam umas às outras, forma-se um tampão plaquetário. À medida que a cascata de coagulação é ativada, a trombina converte o fibrinogénio, mais fraco, em fibrina, mais forte, criando um coágulo muito mais estável.
Imagem por Lecturio.
Adesão plaquetária
A exposição do sangue a componentes subendoteliais no local da lesão faz com que as plaquetas adiram ao local da lesão.
Os recetores de GpIb nas plaquetas ligam-se ao fator de von Willebrand exposto (FvW), dentro da matriz subendotelial. Este vínculo é forte o suficiente para resistir às forças de cisalhamento do sangue que flui.
Outras interações de adesão:
Envolvem colágeno, outros recetores de glicoproteínas e recetores de tirosina cinase
Contribuem para a adesão e ativação das plaquetas
As plaquetas que aderem são ativadas.
Ativação de plaquetas
As plaquetas ativadas ampliam a adesão e agregação plaquetária e estimulam a secreção.
Ativadores de plaquetas:
Ativadores plaquetários potentes:
Trombina: produzida na cascata de coagulação
Colágeno: interage com as plaquetas no local da lesão
A cascata da coagulação representa uma série de reações que, no final, gera um forte coágulo de fibrina polimerizado. Este processo também é conhecido como hemostase secundária.
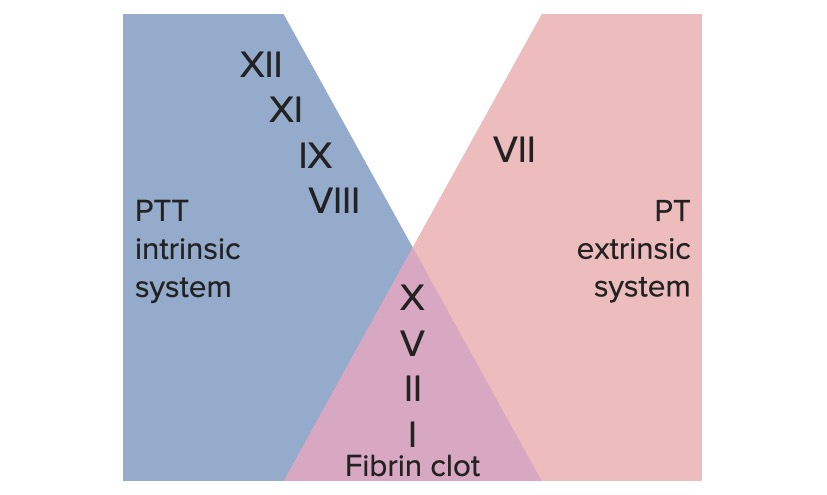
Vários fatores de coagulação sofrem ativação sequencial por 1 das 2 vias:
Via extrínseca: responsável principalmente pela iniciação da cascata
Via intrínseca: envolvida principalmente na amplificação da cascata
Via comum:
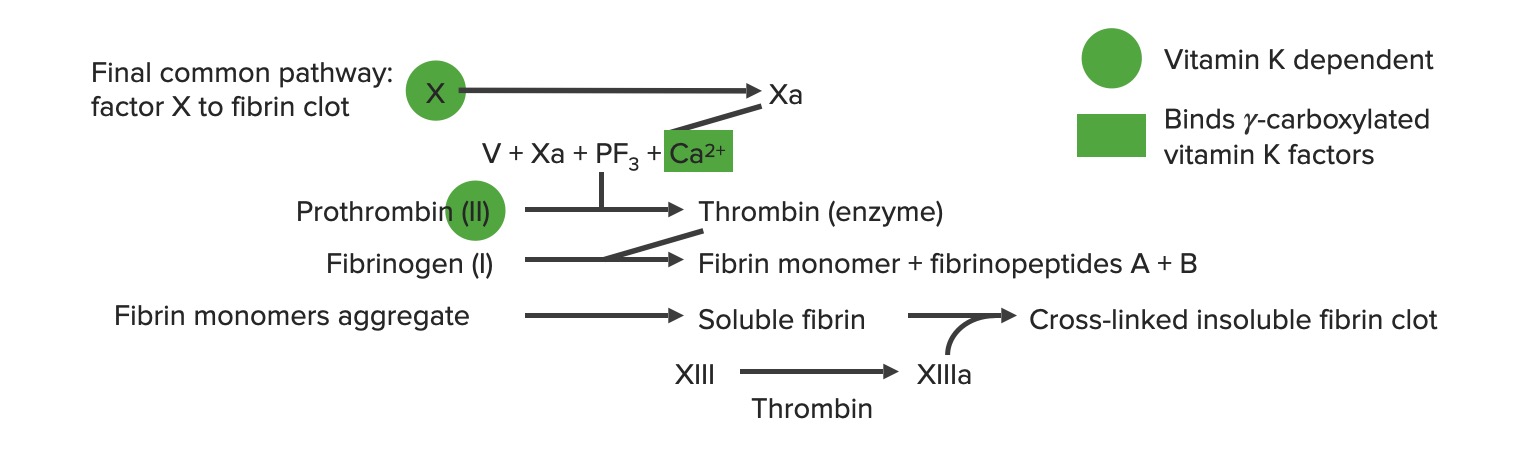
As vias extrínseca e intrínseca unem-se para formar a via comum final, quando o fator X é ativado.
A formação do coágulo de fibrina ocorre no final da via comum.
Iniciação:
A via extrínseca é ativada a partir da lesão endotelial e produz o fator X ativado (Xa).
O fator Xa move-se, então, através da via comum.
A trombina é produzida na via comum.
Amplificação:
A produção inicial de trombina ativa múltiplos fatores nas vias intrínseca e comum.
À medida que a via intrínseca é ativada, é produzida uma quantidade acrescida de fator Xa.
O fator Xa permite o aumento da ativação da via comum:
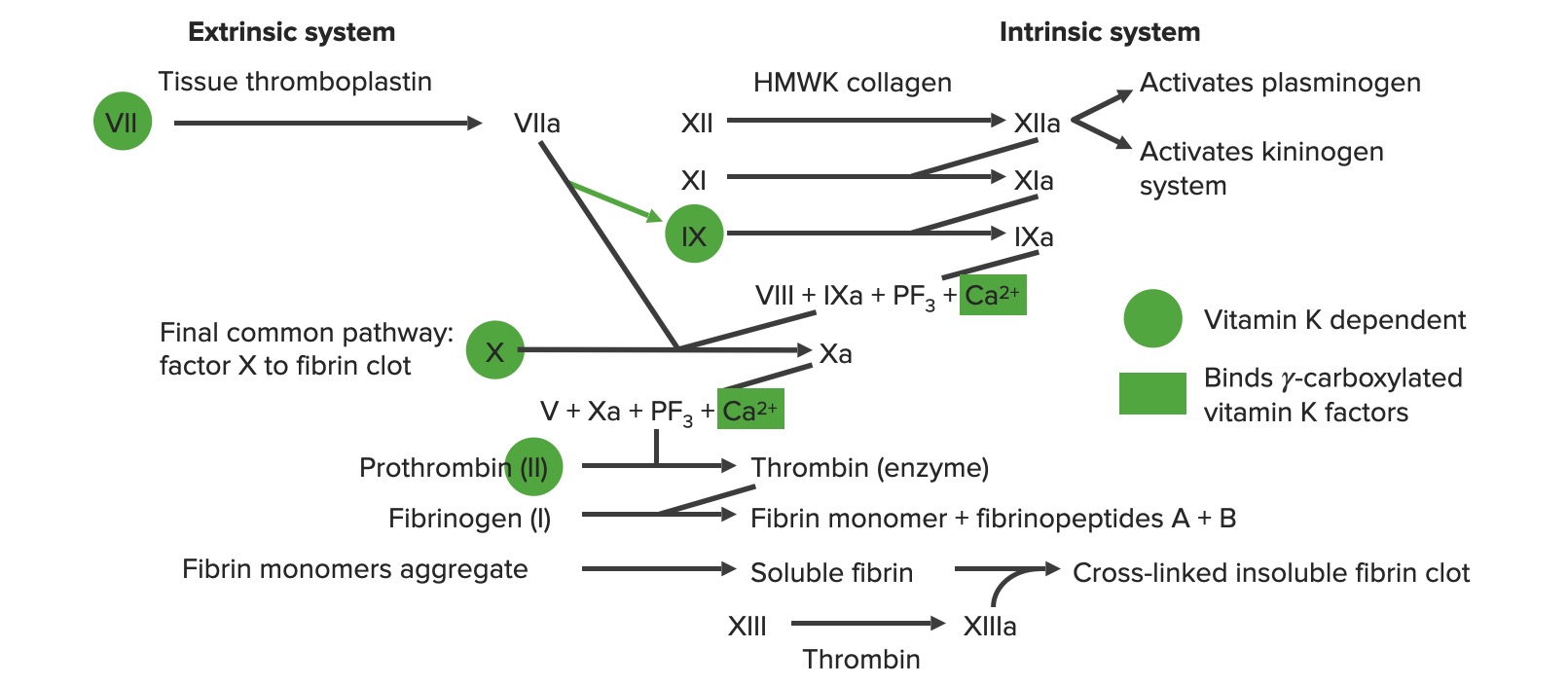
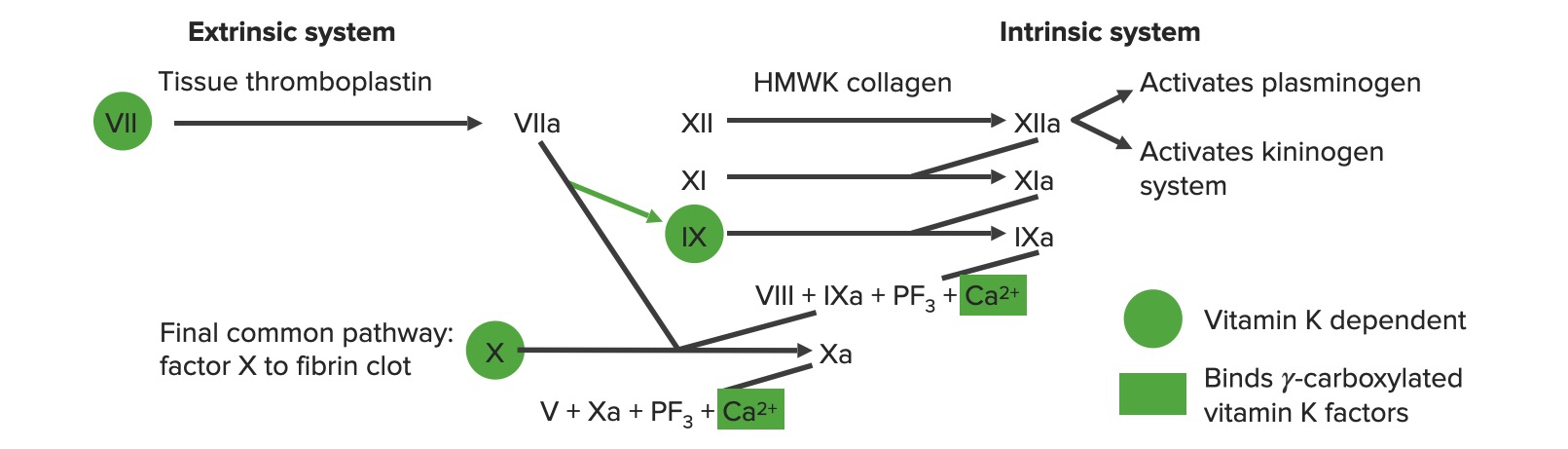
Visão geral da cascata da coagulação a: forma ativada PF3: fator plaquetário 3 (fosfolípidos)
Imagem por Lecturio.
Fatores de coagulação
Os fatores de coagulação são serinoproteases semelhantes à tripsina e são indicados com algarismos romanos.
Todos os fatores pró-coagulantes são sintetizados no fígado, exceto:
Fator VIII: produzido nas células endoteliais
FvW: produzido nos megacariócitos e células endoteliais
Fatores dependentes de vitamina K:
Sofrem carboxilação, para se tornarem funcionais, que requer a presença de vitamina K
Procoagulantes:
Fator II
Fator VII
Fator IX
Fator X
Anticoagulantes:
Proteína C
Proteína S
Vitamina K:
Sintetizada principalmente no cólon
Ativada pela epóxido redutase no fígado
Funciona como um cofator para a gama-glutamil carboxilase para carboxilar os fatores dependentes da vitamina K
Estes fatores carboxilados ganham afinidade pelos fosfolípidos carregados negativamente nas plaquetas → promovem a coagulação
Formam complexos enzimáticos multicomponentes que:
Executam etapas críticas na cascata de coagulação
Cada um contém uma proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS, um cofator e um substrato
Ligados às superfícies membranares de fosfolípidos aniónicos
Restringem a maioria da geração de trombina aos locais de lesão vascular
Fator VIIa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + fator tecidual (cofator) + fator X (substrato)
Ativa o fator X → fator Xa
X-ase intrínseca:
Fator IXa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + fator VIIIa (cofator) + fator X (substrato)
Ativa o fator X → fator Xa
Protrombinase:
Fator Xa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) + fator VaVAVentilation: Mechanics of Breathing (cofator) + protrombina (substrato)
Ativa a protrombina → trombina
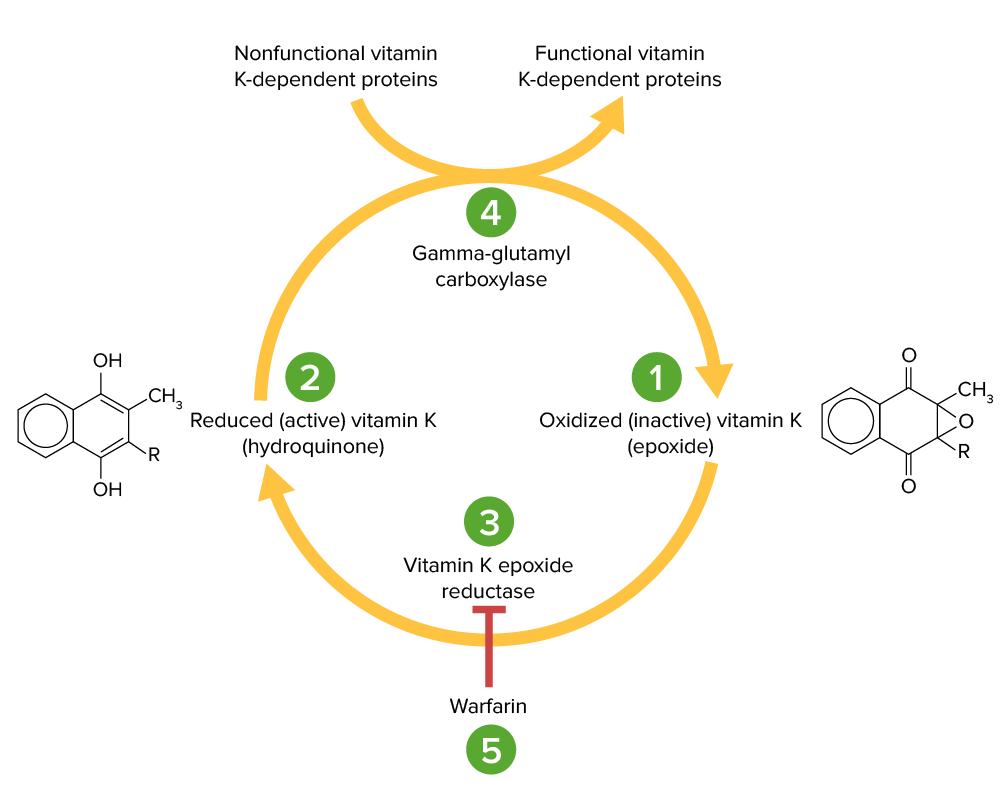
Ciclo da vitamina K: O epóxido de vitamina K (1) é inativo e convertido na sua forma ativa e reduzida, a vitamina K hidroquinona (2), pela vitamina K epóxido redutase (VKOR; 3). A vitamina K hidroquinona é um cofator na carboxilação de resíduos específicos de glutamato nas proteínas dependentes da vitamina K (fatores II, VII, IX, X, proteína C e S), um processo necessário para as ativar. A reação de carboxilação é catalisada pela gama-glutamil carboxilase (4). A vitamina K hidroquinona é oxidada para a forma de epóxido quando atua como cofator, mas é depois reciclada de novo para a forma de hidroquinona pela VKOR. A varfarina inibe a VKOR (5), de modo que a vitamina K não pode ser reciclada da sua forma oxidada para a forma reduzida. Assim, as proteínas dependentes da vitamina K não podem ser ativadas.
Imagem por Lecturio.
Via extrínseca: A via do fator tecidual
A via extrínseca é o mecanismo fisiológico primário pelo qual a coagulação é iniciada.
Envolve X-ase extrínseca
Começa com a exposição do fator tecidual na matriz subendotelial:
Glicoproteína membranar
Expressa apenas após a lesão endotelial
O fator tecidual ativa o fator VII → VIIa
O fator VIIa ativa o fator X → Xa. O fator Xa é o 1º passo na via comum.
Para resumir, o fator tecidual ativa VII → VIIa, que ativa X → Xa → via comum
Via intrínseca: via de contato
A via intrínseca é a principal responsável pela amplificação da ativação do fator X. O fator X é ativado pela trombina inicial, gerada pela via extrínseca/comum, mas também pode ser ativado diretamente pela lesão endotelial.
A exposição do colágeno da matriz subendotelial, carregado negativamente, ativa o cininogénio de alto peso molecular (HMWK, pela sigla em inglês) e a pré-calicreína (PK, pela sigla em inglês).
HMWK + PK ativam o fator XII → XIIa
O Fator XIIa ativa:
Fator XI → XIa
A trombina (da via comum) também ativa o fator XI.
Pré-calicreína → calicreína
A calicreína amplifica a ativação de XII → XIIa
O fator XIa ativa o fator IX → IXa
X-ase intrínseca: o fator IXa (proteaseProteaseEnzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene.HIV Infection and AIDS) associa-se ao fator VIIIa (cofator) para ativar o fator X (substrato) → Xa
Fator VIII:
Ativado pelo fator Xa e trombina (ambos gerados, inicialmente, pelas vias extrínseca e comum)
Estabilizado pelo FvW
O fator Xa é o 1º passo na via comum.
Para resumir, HMWK + PK ativa → 12, que ativa → 11, que ativa → 9, que se associa ao 8 para ativar → 10
Os sistemas de coagulação extrínseca e intrínseca
Imagem por Lecturio.
Via comum
Inicia-se com a protrombinase: o fator Xa combina-se com o fator VaVAVentilation: Mechanics of Breathing e o cálcio para ativar a protrombina (fator II) → trombina (fator IIa)
O corpo produz várias substâncias que inibem a ligação, agregação e secreção plaquetária, e que funcionam como anticoagulantes naturais. Estes mecanismos limitam a coagulação a locais focais específicos e mantêm o sangue fluido.
Inibidor da via do fator tecidual (TFPITFPIHemostasis, pela sigla em inglês):
Inibe a ativação do fator X
Localizado principalmente na superfície das células endoteliais microvasculares
Antitrombina:
Anticoagulante circulante, natural, produzido pelo fígado
Inibe as formas ativadas dos fatores II, IX e X
A taxa de inativação do fator é aumentada pela heparina
Proteínas C e S:
Fatores dependentes de vitamina K, produzidos pelo fígado
A proteína C cliva e inativa os fatores V e VIII.
A proteína S aumenta a atividade da proteína C.
Outras substâncias anticoagulantes produzidas pelas células endoteliais:
Prostaciclina: vasodilatador que bloqueia a agregação plaquetária
Óxido nítrico: vasodilatador que bloqueia a adesão e agregação plaquetária
Trombomodulina: liga-se à trombina e converte-a num anticoagulante que ativa a proteína C
Fase fibrinolítica
O sistema fibrinolítico visa a remoção do coágulo, após a reparação da vasculatura. O processo é realizado principalmente pela plasmina.
Plasmina: cliva os polímeros de fibrina (fibrinólise)
O plasminogénio é ativado (convertido em plasmina) pelo:
Ativador de plasminogénio tecidual (TPatPAIschemic Stroke, pela sigla em inglês)
Ativador do plasminogénio urinário (UPa, pela sigla em inglês) também conhecido como urocinase
Ambos são secretados por células endoteliais.
Fibrinólise:
Forma produtos de degradação da fibrina (e.g., D-dímeros)
Gera novos locais de ligação à plasmina, na fibrina parcialmente degradada
Tempo necessário para o plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products coagular, quando exposto ao fator tecidual
Mede a função das vias extrínseca e comum
Intervalo de referência: aproximadamente 11–13 segundos
Elevado na:
Terapêutica com varfarina
Défice de vitamina K
Défice dos fatores II, V, VII e X
Doença hepática
Coagulação intravascular disseminada (CID)
INR:
Relação que compara o PT do doente com um PT de referência
Mede a função das vias extrínseca e comum
Intervalo de referência: aproximadamente 0,8–1,1
PTT:
Tempo necessário para o plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products coagular, quando exposto a uma substância carregada negativamente (que ativa a via intrínseca)
Mede a função das vias intrínseca e comum
Intervalo de referência: 25–40 seg
Elevado na:
Terapêutica com heparina
Hemofilia (alteração do fator VIII ou IX)
Doença de von Willebrand (DvW)
Doença hepática
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
Tempo de sangramento (BT, pela sigla em inglês):
Mede a função plaquetária
Intervalo de referência: 2-7 minutos
Prolongado na:
Trombocitopenia
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
DvW
Doença de Bernard-Soulier
Trombastenia de Glanzmann
Insuficiência renal
Uso de AINEs e/ou aspirina
Fibrinogénio:
Precursor da fibrina
Níveis anormalmente baixos podem aumentar o risco de hemorragia.
Intervalo de referência: 200–400 mg/dL
Tende a ocorrer sangramento anormal quando os níveis são <100 mg/dL
D-dímeros:
Produto primário da degradação da fibrina
Libertados após a clivagem da fibrina polimerizada, pela plasmina
Indica coagulação e fibrinólise recentes ou em curso
Distúrbios da hemostase primária (formação do tampão plaquetário)
Trombastenia de Glanzmann: síndrome hemorrágica, autossómica recessiva, caracterizada por um défice de recetores GpIIb/IIIa, resultando na ausência de agregação plaquetária
Síndrome de Bernard-Soulier: síndrome hemorrágica, autossómica recessiva, caracterizada pelo défice de recetores GpIb, resultando na falência da adesão plaquetária. A síndrome de Bernard-Soulier pode ser diagnosticada através do teste da ristocetina. A ristocetina ativa o FvW para permitir a ligação ao recetor GpIb das plaquetas; no entanto, na síndrome de Bernard-Soulier, as plaquetas não aderem no ensaio.
Trombocitopenia imune: distúrbio autoimune, caracterizado pela presença de autoanticorpos anti-GpIIb/IIIa, que causam destruição plaquetária. A trombocitopenia imune, geralmente, ocorre após infeções virais respiratórias ou gastrointestinais, embora também possa representar uma condição induzida por fármacos. Clinicamente, a trombocitopenia imune pode apresentar hemorragia prolongada, petéquias, hematomas fáceis e/ou púrpura. O tratamento pode incluir uma transfusão de plaquetas ou esplenectomia, ou tratamento com esteroides e imunoglobulinas IV.
Púrpura trombocitopénica trombótica (PTT): distúrbio hemorrágico marcado por uma pentade de febre, anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica microangiopática, trombocitopenia, insuficiência renal e sintomas neurológicos. A púrpura trombocitopénica trombótica ocorre devido a um défice congénito ou adquirido de ADAMTS-13, uma metaloprotease que cliva o FvW. Um défice de ADAMTS-13 resulta em grandes multímeros de FvW, que aumentam a agregação plaquetária, levando à trombose microvascular e ao consumo de plaquetas.
Distúrbios da hemostase secundária (a cascata de coagulação)
Hemofilia: distúrbio raro da coagulação sanguínea, no qual o corpo não possui fatores de coagulação sanguíneos (fator VIII na hemofilia A; fator IX na hemofilia B). Os indivíduos afetados apresentam hemorragia de forma anormal, que pode ocorrer espontaneamente ou após pequenos traumas. Estes indivíduos podem sangrar nos espaços articulares e desenvolver hemorragias internas com risco de vida.
Distúrbios mistos que afetam tanto as plaquetas quanto os fatores de coagulação
DvW: distúrbio hereditário maisMAISAndrogen Insensitivity Syndrome comum da hemostase, causado por um défice qualitativo ou quantitativo de fator de von Willebrand. Existem 3 tipos primários, que diferem em gravidade, embora todos tendam a apresentar alterações hemorrágicas. O fator de Von Willebrand é necessário tanto para a adesão plaquetária inicial quanto para ajudar a estabilizar o fator VIII, na via intrínseca.
CID: condição médica grave, na qual a cascata de coagulação é ativada sistemicamente, levando a múltiplos coágulos que podem causar danos permanentes em órgãos-alvo. Durante a DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation, os fatores de coagulação são completamente consumidos. A coagulação intravascular disseminada tem sempre uma causa secundária. As infeções, queimaduras e neoplasias estão entre as causas maisMAISAndrogen Insensitivity Syndrome comuns. A coagulação intravascular disseminada pode, também, ocorrer durante uma hemorragia pós-parto grave. Os achados laboratoriais incluem trombocitopenia, prolongamento do PT e PTT e elevação dos níveis de D-dímeros.
Cirrose: O fígado é o principal local de síntese da maioria dos fatores de coagulação. Além de prejudicar a síntese de fatores de coagulação, a cirrose pode, também, resultar de forma independente em trombocitopenia, devido ao sequestro plaquetário esplénico e à diminuição da produção de trombopoietina. As próprias plaquetas também podem ser disfuncionais.
Distúrbios da fibrinólise
Mutação do fator V Leiden: resulta na produção de fator V mutado, resistente à degradação pela proteína C ativada, levando assim ao aumento da produção de trombina e a um estado pró-coagulante. As complicações incluem trombose venosa profunda, trombose venosa cerebral e embolia pulmonar.
Mutação dogeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics da protrombina: segunda trombofilia hereditária maisMAISAndrogen Insensitivity Syndrome comum, após o fator V Leiden. Mutações pontuais no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics da protrombina levam ao aumento dos níveis de protrombina, levando a um estado de hipercoagulabilidade e aumento do risco de tromboembolismo venoso.
Deficiência da proteína C ou S: resulta na ausência de inativação dos fatores VaVAVentilation: Mechanics of Breathing e VIIIa. Assim como no fator V Leiden, há um risco aumentado de tromboembolismo venoso e necrose cutânea induzida pela varfarina.
Deficiência de antitrombina: distúrbio hereditário ou adquirido, que resulta na redução da atividade da antitrombina para < 80% da sua atividade normal. A deficiência de antitrombina leva a uma diminuição da inibição dos fatores II, IX e X, criando assim um estado de hipercoagulabilidade.
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.