An essential amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids is an amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids that must come from the diet. Alternatively, a nonessential amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids can be produced by cells and does not require dietary intake. Various substrates undergo a series of processes to make amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids. There are 11 amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids that can be completely synthesized; the other 9 are considered essential and must be included in an individual’s diet. Nonessential amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids are alanine, arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle, asparagine, aspartic acid, cysteine, glutamic acidGlutamic acidA non-essential amino acid naturally occurring in the l-form. Glutamic acid is the most common excitatory neurotransmitter in the central nervous system.Urea Cycle, glutamine, glycine, proline, serine, and tyrosine. Deficiency in the enzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes required for amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids metabolism lead to serious conditions that often present early in life, such as maple syrup urine diseaseMaple syrup urine diseaseAn autosomal recessive inherited disorder with multiple forms of phenotypic expression, caused by a defect in the oxidative decarboxylation of branched-chain amino acids. These metabolites accumulate in body fluids and render a 'maple syrup' odor. The disease is divided into classic, intermediate, intermittent, and thiamine responsive subtypes. The classic form presents in the first week of life with ketoacidosis, hypoglycemia, emesis, neonatal seizures, and hypertonia. The intermediate and intermittent forms present in childhood or later with acute episodes of ataxia and vomiting.Disorders of Amino Acid Metabolism and phenylketonuria.
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids are building blocks for proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis and metabolic intermediates for reactions. Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids are classified as essential or nonessential. Essential amino acidsEssential amino acidsAmino acids that are not synthesized by the human body in amounts sufficient to carry out physiological functions. They are obtained from dietary foodstuffs.Basics of Amino Acids must be incorporated into the diet because the body cannot produce them at sufficient levels to meet physiologic demands; nonessential amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids can be produced by the body.
Essential amino acidsEssential amino acidsAmino acids that are not synthesized by the human body in amounts sufficient to carry out physiological functions. They are obtained from dietary foodstuffs.Basics of Amino Acids include:
Phenylalanine
Valine
Threonine
Tryptophan
Methionine
Leucine
Isoleucine
Lysine
Histidine
Nonessential amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids include:
Alanine
ArginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle
Asparagine
Aspartic acid (aspartate)
Cysteine
Glutamine
Glutamic acidGlutamic acidA non-essential amino acid naturally occurring in the l-form. Glutamic acid is the most common excitatory neurotransmitter in the central nervous system.Urea Cycle (glutamate)
Glycine
Proline
Serine
Tyrosine
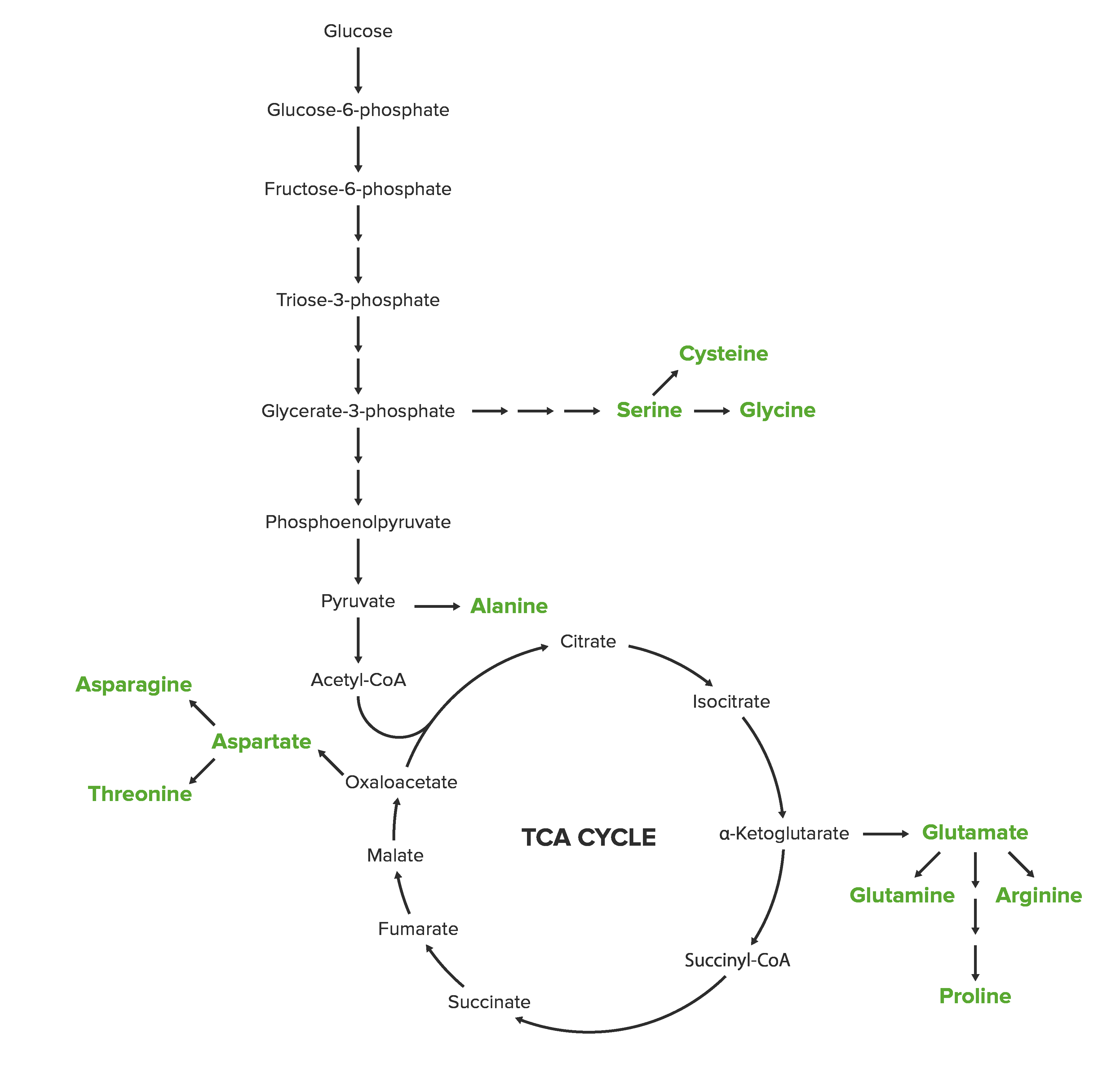
Amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino AcidssynthesisSynthesisPolymerase Chain Reaction (PCR) reactions can be grouped by biosynthetic families, named according to their common starting metabolite.
Biosynthetic families
Table: Comparison of biosynthetic families
Biosynthetic family
Amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids
OxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle
PhosphoenolpyruvatePhosphoenolpyruvateA monocarboxylic acid anion derived from selective deprotonation of the carboxy group of phosphoenolpyruvic acid. It is a metabolic intermediate in glycolysis; gluconeogenesis; and other pathways.Glycolysis & erythrose 4-phosphate
*Essential amino acids that must be incorporated into the diet. Production of these amino acids commonly occurs in prokaryotic organisms and plants. AA: amino acid
Glutamate, glutamine, proline, and arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle are synthesized from α-ketoacids, which are produced by the citric acid cycleCitric acid cycleThe citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or the Krebs cycle, is a cyclic set of reactions that occurs in the mitochondrial matrix. The TCA cycle is the continuation of any metabolic pathway that produces pyruvate, which is converted into its main substrate, acetyl-CoA. Citric Acid Cycle.
TransaminationTransaminationTransamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AA with an alpha-keto group (=O) instead of an alpha-amino group (NH2).Catabolism of Amino Acids is an essential reaction in the production of these amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids.
The enzyme involved in this reaction is an aminotransferase.
Glutamate serves as a precursor for the production of many amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids, such as glutamine, proline, and arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle.
α-ketoacid + glutamate ⇄ amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids + α-ketoglutarate
Glutamate itself is formed by amination of α-ketoglutarate: α-ketoglutarate + NH4+ ⇄ glutamate
Glutamate is produced from α-ketoglutarate through the process of amination.
α-ketoglutarate + NH4+ ⇄ glutamate
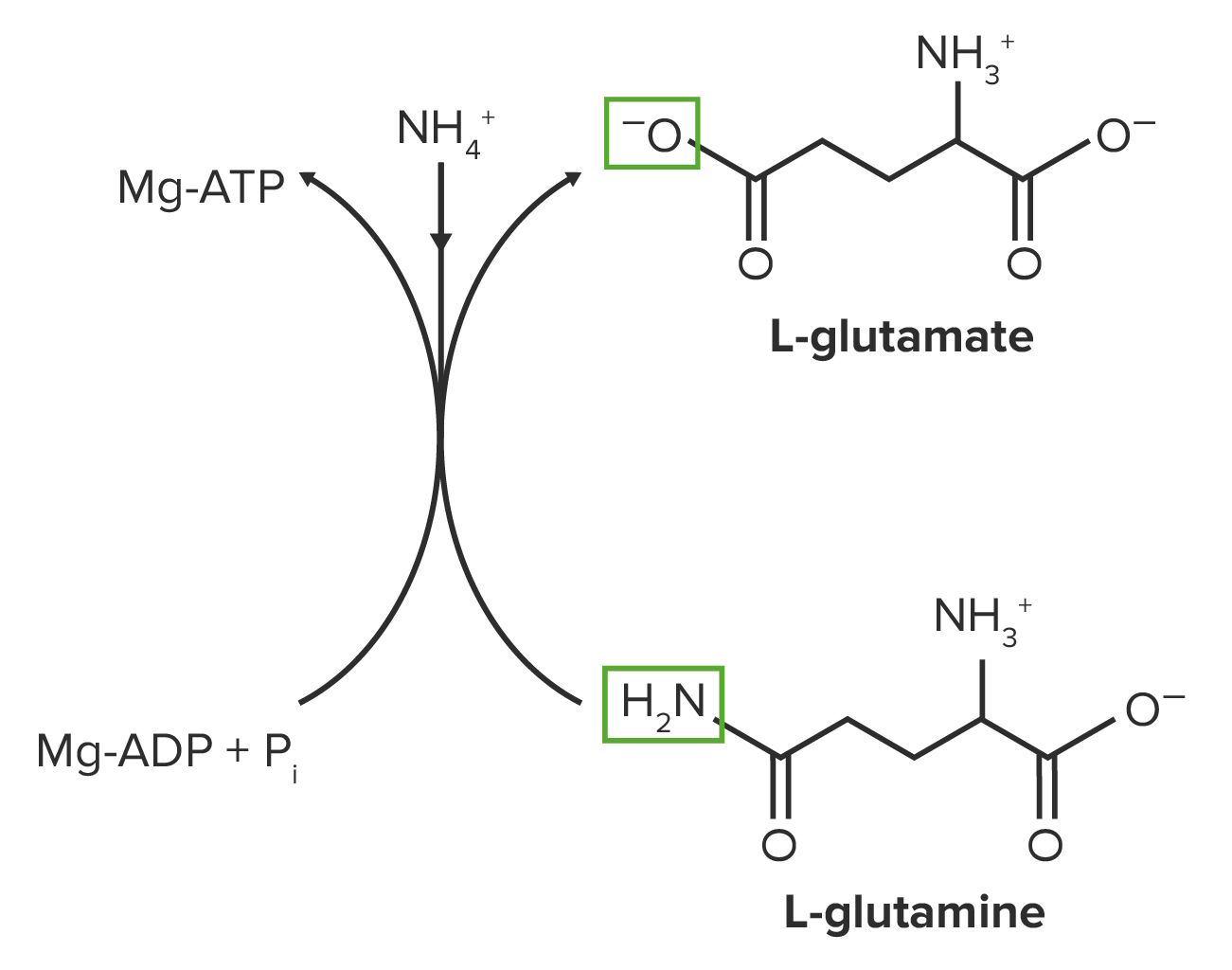
Glutamate serves as the primary precursor for glutamine, proline, and arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle.
Glutamate is converted to glutamine by glutamine synthetase.
Glutamine synthetase uses:
ATP
NH4+: helps prevent ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance build-up
Glutamine synthetase inhibitors:
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids: glycine, alanine, serine, histidine, tryptophan
NucleotidesNucleotidesThe monomeric units from which DNA or RNA polymers are constructed. They consist of a purine or pyrimidine base, a pentose sugar, and a phosphate group.Nucleic Acids: AMP, cytidine triphosphate (CTPCTPPurine and Pyrimidine Metabolism)
Other: carbamoyl phosphatePhosphateInorganic salts of phosphoric acid.Electrolytes, glucosamine-6-phosphate
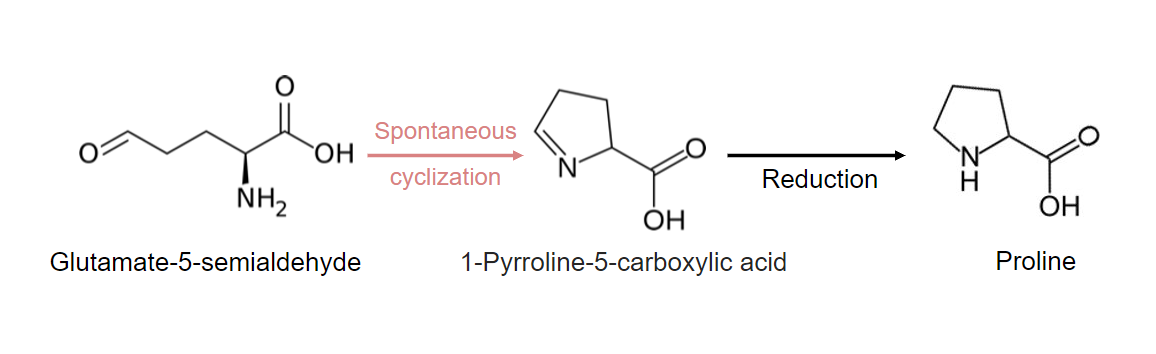
Catalyzed by glutamate-5-kinase and glutamate-5-semialdehyde dehydrogenase
Requires ATP and nicotinamide adenine dinucleotideNicotinamide adenine dinucleotideA coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH).Pentose Phosphate Pathway (NADH)
Produces glutamate-5-semialdehyde
Oxidation and dephosphorylation:
Spontaneous cyclization reaction
Produces 1-pyrroline-5-carboxylic acid
Reduction:
Catalyzed by pyrroline-5-carboxylate reductase
Requires NADH
Generates proline
Cyclization and reduction reactions contributing to proline synthesis: These steps require ATP, NADH, and a cyclization intermediate.
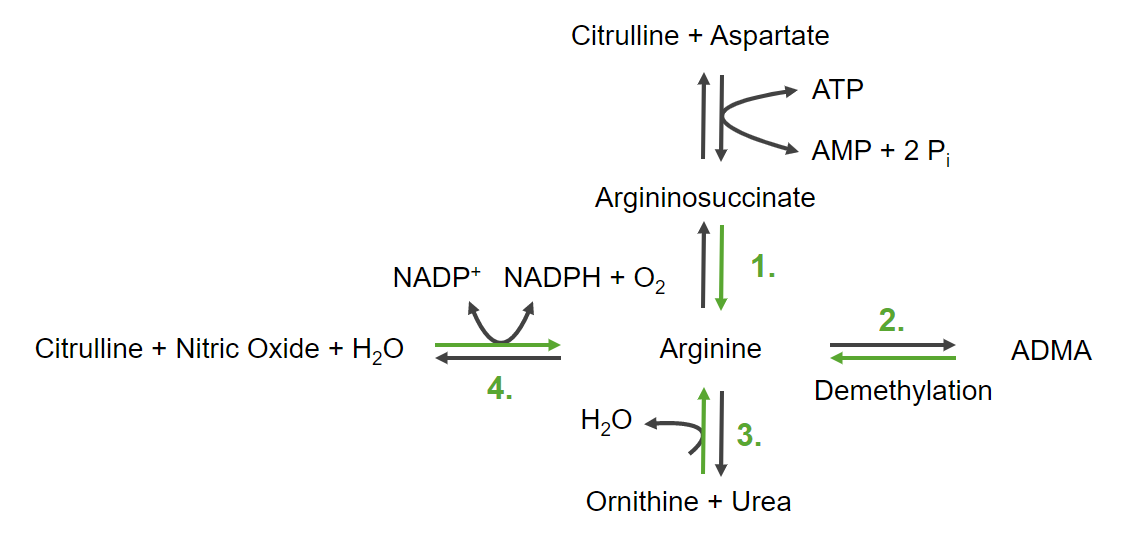
ArgininosuccinateArgininosuccinateThis amino acid is formed during the urea cycle from citrulline, aspartate and ATP. This reaction is catalyzed by argininosuccinic acid synthetase.Urea Cycle serves as an intermediate compound.
Reversible demethylation from asymmetric dimethylarginine (ADMA)
Reversible reaction from ornithineOrnithineAn amino acid produced in the urea cycle by the splitting off of urea from arginine.Urea Cycle and ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle
Reversible reaction from citrullineCitrullineUrea Cycle, nitric oxideNitric OxideA free radical gas produced endogenously by a variety of mammalian cells, synthesized from arginine by nitric oxide synthase. Nitric oxide is one of the endothelium-dependent relaxing factors released by the vascular endothelium and mediates vasodilation. It also inhibits platelet aggregation, induces disaggregation of aggregated platelets, and inhibits platelet adhesion to the vascular endothelium. Nitric oxide activates cytosolic guanylate cyclase and thus elevates intracellular levels of cyclic gmp.Pulmonary Hypertension Drugs, and nicotinamide adenine dinucleotideNicotinamide adenine dinucleotideA coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH).Pentose Phosphate Pathway (NADP+)
The 4 ways to produce arginine ADMA: asymmetric dimethylarginine NADPH and NADP+: nicotinamide adenine dinucleotide Pi: inorganic phosphate
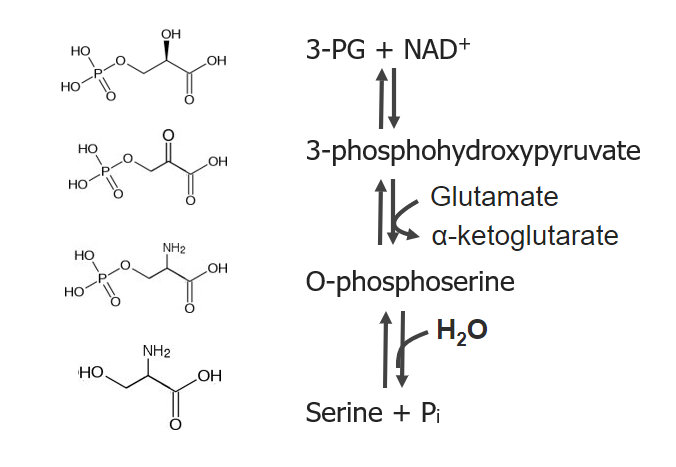
Serine, glycine, and cysteine are amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids included in the 3-phosphoglycerate3-phosphoglycerateGlycolysis (3-PG) biosynthetic family. Serine is the first amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids produced in this group, and it subsequently contributes to the production of glycine and cysteine.
TransaminationTransaminationTransamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AA with an alpha-keto group (=O) instead of an alpha-amino group (NH2).Catabolism of Amino Acids of 3-phosphohydroxypyruvate
Catalyzed by phosphoserine transaminaseTransaminaseA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Catabolism of Amino Acids
Produces O-phosphoserine
HydrolysisHydrolysisThe process of cleaving a chemical compound by the addition of a molecule of water.Proteins and Peptides of O-phosphoserine
Catalyzed by phosphoserine phosphatase
Produces serine
Serine is made via 3 reactions, beginning with 3-phosphoglycerate (PG) and nicotinamide adenine dinucleotide (NAD+)
Aspartate, lysine, threonine, asparagine, methionine, and isoleucine are amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids that have oxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle as a common precursor.
Aspartate and asparagine are the only nonessential amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids in this family.
OxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle is first converted to aspartate.
Aspartate is then converted to asparagine.
Aspartate
Aspartate is produced by a transaminationTransaminationTransamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AA with an alpha-keto group (=O) instead of an alpha-amino group (NH2).Catabolism of Amino Acids reaction.
Catalyzed by an aminotransferase: transfers an amine group from another amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids
Produces aspartate from oxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle
Asparagine
Asparagine is also produced by a transaminationTransaminationTransamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AA with an alpha-keto group (=O) instead of an alpha-amino group (NH2).Catabolism of Amino Acids reaction.
Alanine, valine, and leucine share pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis as a common precursor. Alanine is the only amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids in the pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis biosynthetic family that can be produced by humans.
PyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis is an end product of glycolysisGlycolysisGlycolysis is a central metabolic pathway responsible for the breakdown of glucose and plays a vital role in generating free energy for the cell and metabolites for further oxidative degradation. Glucose primarily becomes available in the blood as a result of glycogen breakdown or from its synthesis from noncarbohydrate precursors (gluconeogenesis) and is imported into cells by specific transport proteins. Glycolysis.
Feedback inhibition is provided by the final product (e.g., alanine).
Alanine
Alanine is produced through a 2-step reaction.
α-ketoglutarate is converted to glutamate:
Catalyzed by glutamate dehydrogenase
Requires ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance and NADH
Glutamate transfers an amino group to pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis to form alanine:
Catalyzed by an aminotransferase enzymeAminotransferase enzymeA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Catabolism of Amino Acids
Requires preformed pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis from glycolysisGlycolysisGlycolysis is a central metabolic pathway responsible for the breakdown of glucose and plays a vital role in generating free energy for the cell and metabolites for further oxidative degradation. Glucose primarily becomes available in the blood as a result of glycogen breakdown or from its synthesis from noncarbohydrate precursors (gluconeogenesis) and is imported into cells by specific transport proteins. Glycolysis
Phosphoenolpyruvate Biosynthetic Family
Overview
The aromatic amino acidsAromatic amino acidsAmino acids containing an aromatic side chain.Basics of Amino Acids phenylalanine, tryptophan, and tyrosine are included in the phosphoenolpyruvatePhosphoenolpyruvateA monocarboxylic acid anion derived from selective deprotonation of the carboxy group of phosphoenolpyruvic acid. It is a metabolic intermediate in glycolysis; gluconeogenesis; and other pathways.Glycolysis family. Phenylalanine and tryptophan are essential amino acidsEssential amino acidsAmino acids that are not synthesized by the human body in amounts sufficient to carry out physiological functions. They are obtained from dietary foodstuffs.Basics of Amino Acids; tyrosine is a nonessential amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids.
Tyrosine
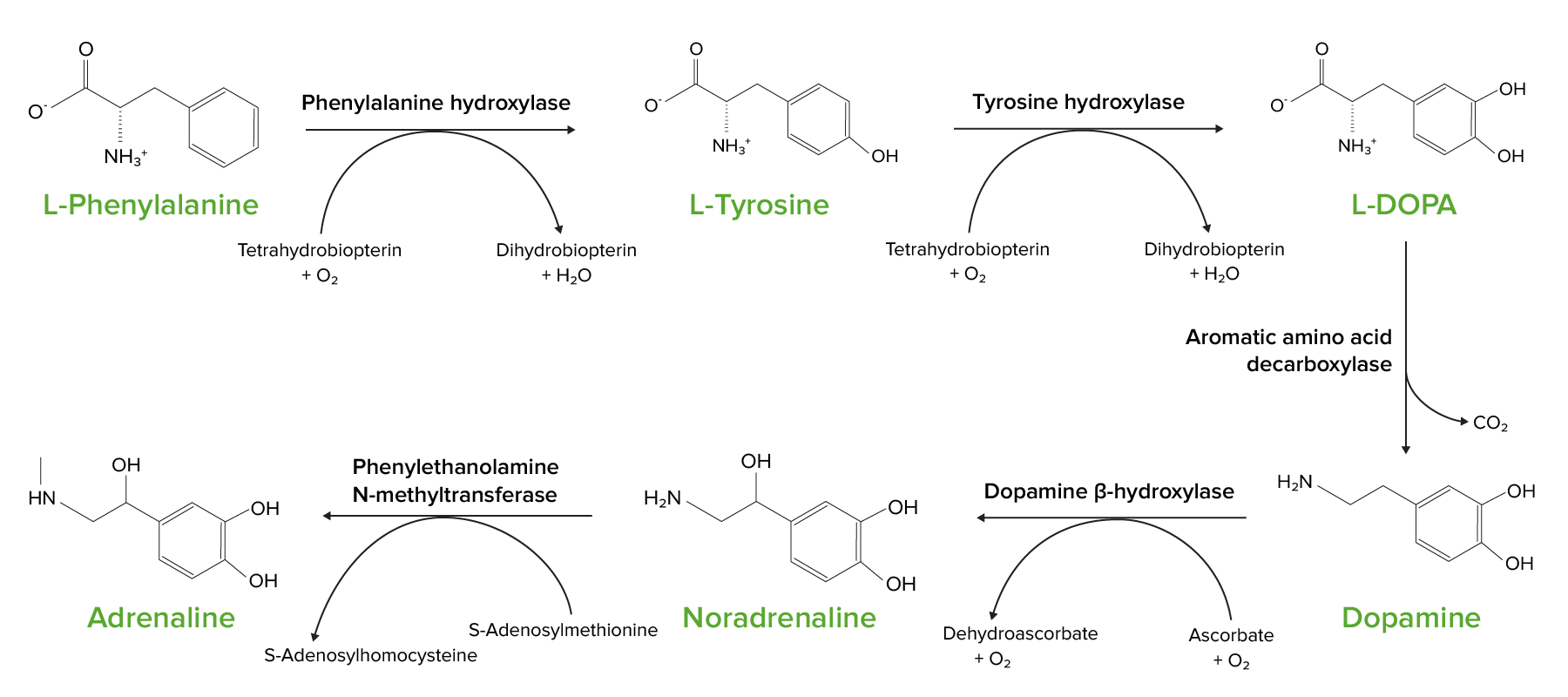
Phenylalanine serves as the precursor for tyrosine
The reaction is catalyzed by biopterin-dependent phenylalanine hydroxylase.
Phenylalanine hydroxylase deficiency results in phenylketonuria.
Tyrosine is the precursor for important neurotransmitters:
DopamineDopamineOne of the catecholamine neurotransmitters in the brain. It is derived from tyrosine and is the precursor to norepinephrine and epinephrine. Dopamine is a major transmitter in the extrapyramidal system of the brain, and important in regulating movement.Receptors and Neurotransmitters of the CNS
Noradrenaline
Adrenaline
The conversion of L-phenylalanine into L-tyrosine is the 1st step in the upper left. Tyrosine and phenylalanine are precursors to a number of crucial neurotransmitters and hormones.
Phenylketonuria (PKU): defect of phenylalanine hydroxylase that results in impairment of the conversion of phenylalanine to tyrosine and subsequent accumulation of phenylalanine. Individuals will present with psychomotor delay and seizures, and their sweat will classically have a “mousy” odor. It is critical for these individuals to avoid ingestion of phenylalanine.
Maple syrup urine diseaseMaple syrup urine diseaseAn autosomal recessive inherited disorder with multiple forms of phenotypic expression, caused by a defect in the oxidative decarboxylation of branched-chain amino acids. These metabolites accumulate in body fluids and render a ‘maple syrup’ odor. The disease is divided into classic, intermediate, intermittent, and thiamine responsive subtypes. The classic form presents in the first week of life with ketoacidosis, hypoglycemia, emesis, neonatal seizures, and hypertonia. The intermediate and intermittent forms present in childhood or later with acute episodes of ataxia and vomiting.Disorders of Amino Acid Metabolism: defect in the branched-chain α-ketoacid dehydrogenase complex that results in the accumulation of branched-chain amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids. Individuals present with cognitive disabilities, sweet-smelling urine, and dystoniaDystoniaDystonia is a hyperkinetic movement disorder characterized by the involuntary contraction of muscles, resulting in abnormal postures or twisting and repetitive movements. Dystonia can present in various ways as may affect many different skeletal muscle groups. Dystonia. The primary treatment is avoidance of branched-chain amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids. Severe cases may require a liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy transplant.
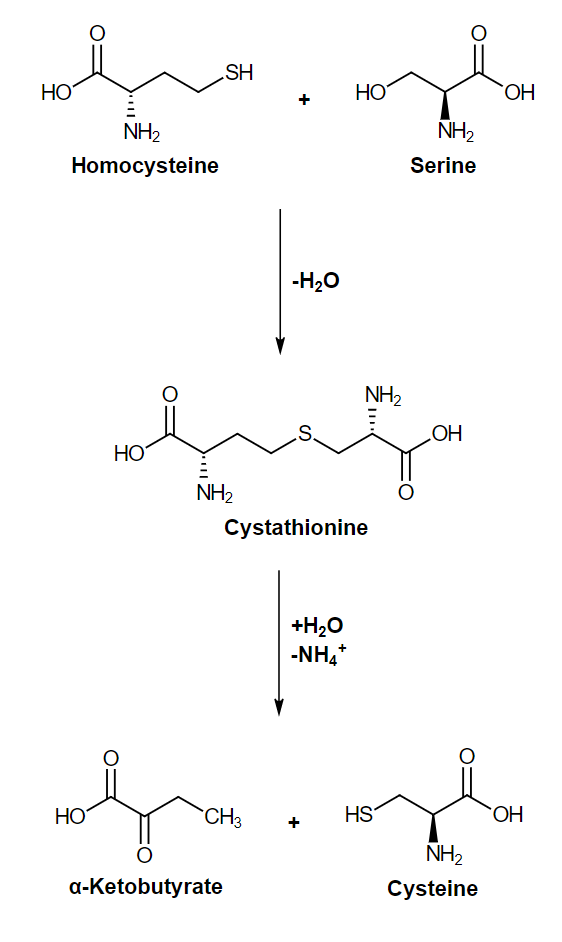
Homocystinuria: defect in the enzyme cystathionine β-synthase, which leads to an accumulation of homocysteine. Individuals present with flushing, developmental delay, downward lensLensA transparent, biconvex structure of the eye, enclosed in a capsule and situated behind the iris and in front of the vitreous humor (vitreous body). It is slightly overlapped at its margin by the ciliary processes. Adaptation by the ciliary body is crucial for ocular accommodation.Eye: Anatomy dislocation, vascular disease, and osteoporosisOsteoporosisOsteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis. It is recommended that these individuals maintain a diet low in sulfur.
Alkaptonuria: deficiency of homogentisic acid dioxygenase, which impairs the normal degradation of tyrosine to fumarateFumarateCitric Acid Cycle. Individuals present with a bluish-black discoloration of connective tissues, arthritisArthritisAcute or chronic inflammation of joints.Osteoarthritis, and calcifications of various tissues. There is currently no treatment for alkaptonuria, but the life expectancyLife expectancyBased on known statistical data, the number of years which any person of a given age may reasonably expected to live.Population Pyramids remains normal in these individuals.
References
Ling, Z.-N., Chen, H.-F., Zhai, X.-L., Pang, W., Jin, S., Kuang, Y., Shao, Y.-W., & Liu, Y.-P. (2023). Amino acid metabolism in health and disease. Signal Transduction and Targeted Therapy, 8(1), 1–32. https://doi.org/10.1038/s41392-023-01569-3
Nelson, D. L., & Cox, M. M. (2021). Lehninger principles of biochemistry (8th ed.). W.H. Freeman.
Sannino, S., Guerrero-Zotano, A., Straubel, J., Balaji, S. A., Zou, Q., Hu, Y., He, J., Kim, S. B., Martin, N. E., Mills, G. B., & Wulf, G. M. (2023). Non-essential amino acid availability influences proteostasis and breast cancer cell survival during proteotoxic stress. Molecular Cancer Research, 21(7), 675–690. https://doi.org/10.1158/1541-7786.MCR-22-0843
Xia, H., Wu, C., Li, H., Li, Z., Yang, Y., Zou, C., Kuang, X., & Kuang, M. (2024). Amino acids and their roles in tumor immunotherapy of breast cancer. The Journal of Gene Medicine, 26(1), e3647. https://doi.org/10.1002/jgm.3647
Zhang, J., Pavlova, N. N., & Thompson, C. B. (2017). Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. The EMBO Journal, 36(10), 1302–1315. https://doi.org/10.15252/embj.201696151
Create your free account or log in to continue reading!