Amyloidosis is a disease caused by abnormal extracellular tissue deposition of fibrils composed of various misfolded low-molecular-weight protein subunits. These proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis are frequently byproducts of other pathological processes (e.g., multiple myeloma). These misfolded proteins Misfolded Proteins Cell Injury and Death can become deposited in different tissues, interfere with normal organ functions, and cause tissue-specific diseases (e.g., renal amyloidosis Renal Amyloidosis Ankylosing Spondylitis causes proteinuria Proteinuria The presence of proteins in the urine, an indicator of kidney diseases. Nephrotic Syndrome in Children). Diagnosis is established clinically and confirmed with tissue biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma. Treatment should be directed toward the underlying cause and the reduction of amyloid deposition.
Last updated: Dec 15, 2025
Amyloidosis is a disease caused by abnormal extracellular tissue deposition of fibrils composed of various misfolded low-molecular-weight protein subunits.
Systemic amyloidosis:
Localized amyloidosis:
Hereditary amyloidosis:
| Disease | Amyloid protein | Organs involved | Specifics |
|---|---|---|---|
| Primary amyloidosis | AL | Systemic involvement:
|
|
| Secondary amyloidosis | AA | ||
| Hemodialysis-associated amyloidosis | Aβ₂M |
|
|
| Disease | Amyloid protein | Organs involved | Specifics |
|---|---|---|---|
| Cerebral amyloidosis | Aβ amyloid |
|
|
| Medullary carcinoma of the thyroid Thyroid The thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck. Thyroid Gland: Anatomy | ACal | Thyroid Thyroid The thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck. Thyroid Gland: Anatomy gland | Clinically insignificant |
| Type 2 diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus | AIAPP | Pancreatic islets of Langerhans | Associated with insulinoma Insulinoma A benign tumor of the pancreatic beta cells. Insulinoma secretes excess insulin resulting in hypoglycemia. Pancreatic Neuroendocrine Tumors (PanNETs) |
| Isolated atrial amyloidosis | AANP | Atria | Associated with aging |
| Disease | Amyloid protein | Organs involved | Specifics |
|---|---|---|---|
| Familial Mediterranean fever Mediterranean fever Brucellosis (also known as undulant fever, mediterranean fever, or malta fever) is a zoonotic infection that spreads predominantly through ingestion of unpasteurized dairy products or direct contact with infected animal products. Clinical manifestations include fever, arthralgias, malaise, lymphadenopathy, and hepatosplenomegaly. Brucella/Brucellosis | AA |
|
|
| Familial amyloid neuropathy Neuropathy Leprosy | ATTR | Peripheral nerves Peripheral Nerves The nerves outside of the brain and spinal cord, including the autonomic, cranial, and spinal nerves. Peripheral nerves contain non-neuronal cells and connective tissue as well as axons. The connective tissue layers include, from the outside to the inside, the epineurium, the perineurium, and the endoneurium. Nervous System: Histology |
|
| Familial amyloid cardiomyopathy Cardiomyopathy Cardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types | ATTR | Ventricles |
|
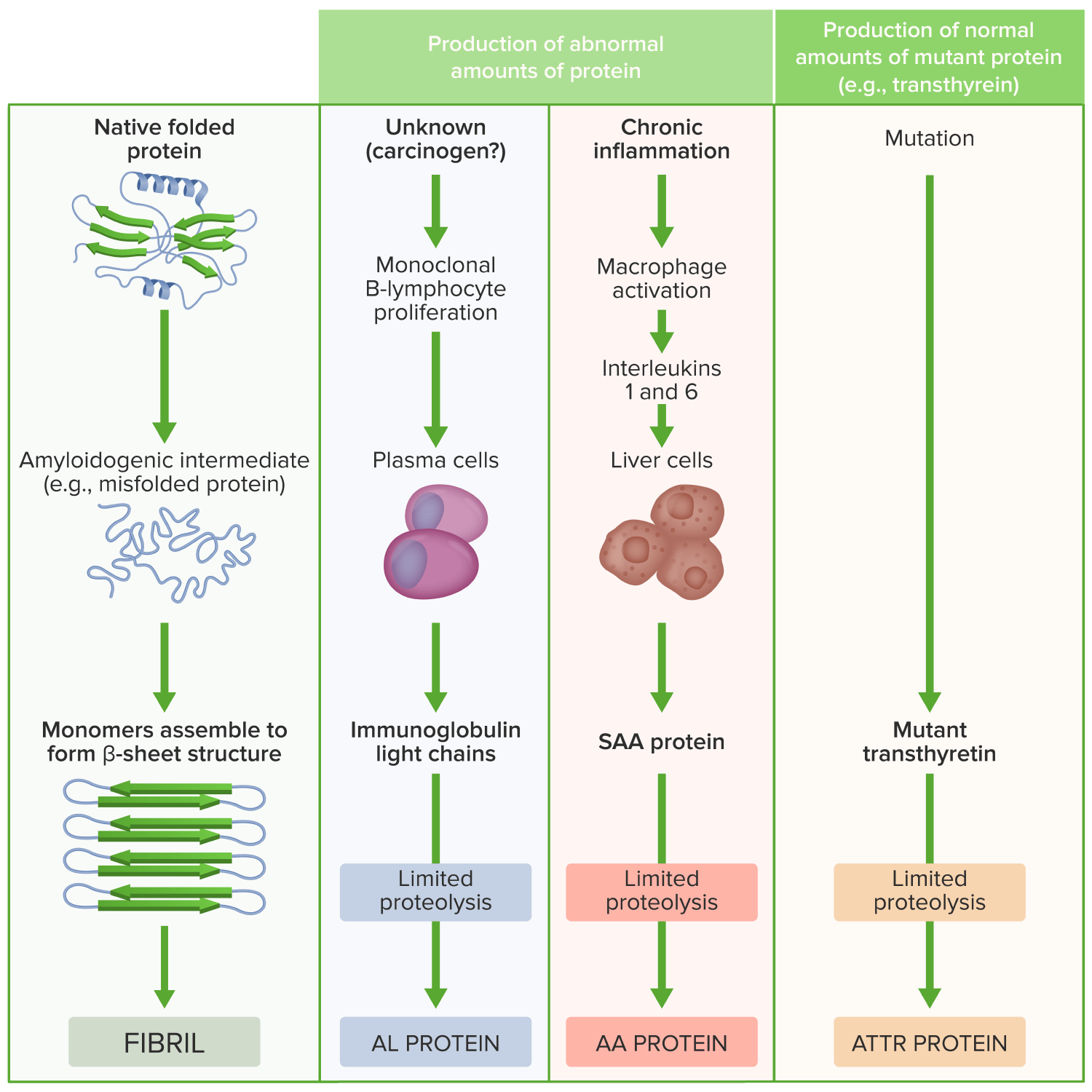
Pathogenesis of amyloidosis
Image by Lecturio.Clinical presentation depends on the organ/organs involved.
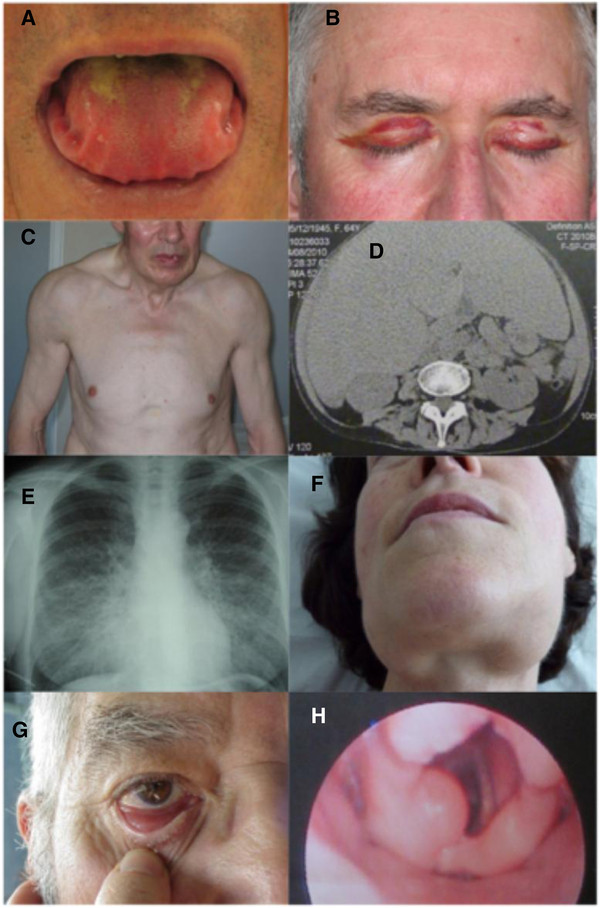
Systemic manifestations of AL amyloidosis:
A: macroglossia with lateral scalloping of the tongue
B: bilateral periorbital purpura
C: pseudo-athletic appearance secondary to diffuse muscular infiltration
D: voluminous hepatomegaly due to primary hepatic amyloidosis
E: diffuse bilateral interstitial lung disease
F: submandibular gland enlargement
Localized AL amyloidosis:
G: nodular conjunctival amyloidosis
H: laryngeal supraglottic amyloid lump
History and physical exam:
Tissue biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma:
Tests specific to the underlying disease:
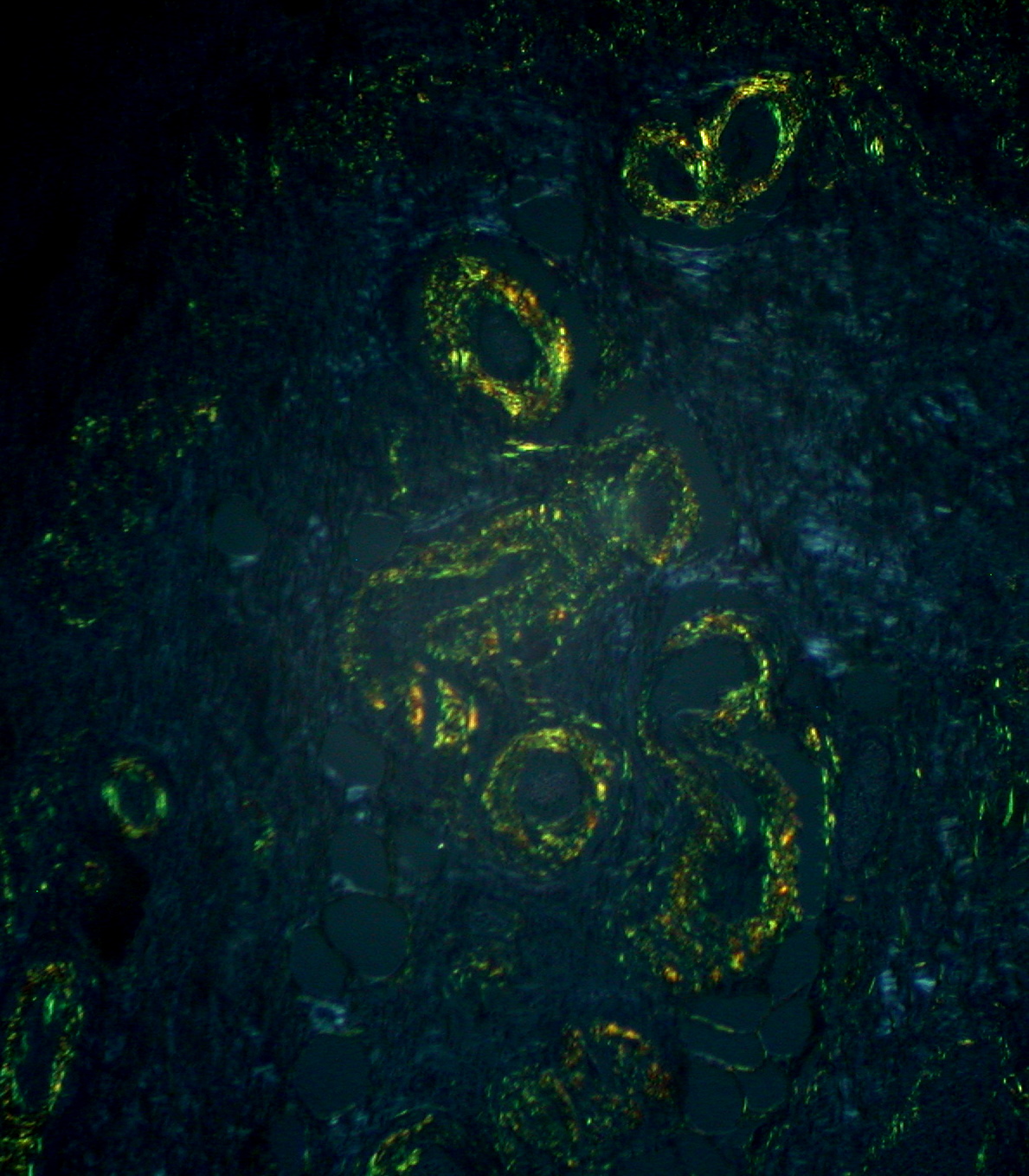
Amyloid: typical apple-green birefringence with polarized light microscopy after Congo red staining
Image: “Gastric Amyloidosis (Congo red stain, crossed polarizers)” by Ed Uthman. License: CC BY 2.0
Gastric amyloidosis: red-colored depositions seen with H&E staining around the vessels (containing RBCs)
Image: “Gastric Amyloidosis (Congo Red Stain)” by Ed Uthman. License: CC BY 2.0