Transmissible spongiform encephalopathies are diseases caused by prions Prions Small proteinaceous infectious particles which resist inactivation by procedures that modify nucleic acids and contain an abnormal isoform of a cellular protein which is a major and necessary component. The abnormal (scrapie) isoform is prpsc (prpsc proteins) and the cellular isoform prpc (prpc proteins). The primary amino acid sequence of the two isoforms is identical. Human diseases caused by prions include creutzfeldt-jakob syndrome; gerstmann-straussler syndrome; and insomnia, fatal familial. Virology. Prions Prions Small proteinaceous infectious particles which resist inactivation by procedures that modify nucleic acids and contain an abnormal isoform of a cellular protein which is a major and necessary component. The abnormal (scrapie) isoform is prpsc (prpsc proteins) and the cellular isoform prpc (prpc proteins). The primary amino acid sequence of the two isoforms is identical. Human diseases caused by prions include creutzfeldt-jakob syndrome; gerstmann-straussler syndrome; and insomnia, fatal familial. Virology differ from viruses Viruses Minute infectious agents whose genomes are composed of DNA or RNA, but not both. They are characterized by a lack of independent metabolism and the inability to replicate outside living host cells. Virology in that they are small, infectious pathogens that do not contain nucleic acid. Recognized spongiform encephalopathies include Creutzfeldt-Jakob Disease (CJD), variant Creutzfeldt-Jakob Disease (vCJD), Kuru, fatal familial insomnia Insomnia Insomnia is a sleep disorder characterized by difficulty in the initiation, maintenance, and consolidation of sleep, leading to impairment of function. Patients may exhibit symptoms such as difficulty falling asleep, disrupted sleep, trouble going back to sleep, early awakenings, and feeling tired upon waking. Insomnia (FFI), and Gerstmann-Straussler syndrome (GSS). Common characteristics of these diseases include dementia Dementia Major neurocognitive disorders (NCD), also known as dementia, are a group of diseases characterized by decline in a person's memory and executive function. These disorders are progressive and persistent diseases that are the leading cause of disability among elderly people worldwide. Major Neurocognitive Disorders, ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia, and myoclonus Myoclonus Involuntary shock-like contractions, irregular in rhythm and amplitude, followed by relaxation, of a muscle or a group of muscles. This condition may be a feature of some central nervous system diseases; (e.g., epilepsy-myoclonic). Nocturnal myoclonus is the principal feature of the nocturnal myoclonus syndrome. Neurological Examination. Unfortunately, these diseases are associated with long incubation Incubation The amount time between exposure to an infectious agent and becoming symptomatic. Rabies Virus periods (20+ years) and once symptoms occur, rapidly progress to death.
Last updated: Dec 15, 2025
Spongiform encephalopathies are extremely rare.
Known human diseases:
The most common spongiform encephalopathy Encephalopathy Hyper-IgM Syndrome is Creutzfeldt-Jakob disease. The variations are:
High-risk factors are:
Prion diseases occur when a normal, ⍺-helical protein known as PrPc is converted into an abnormal, β-pleated protein known as PrPsc.
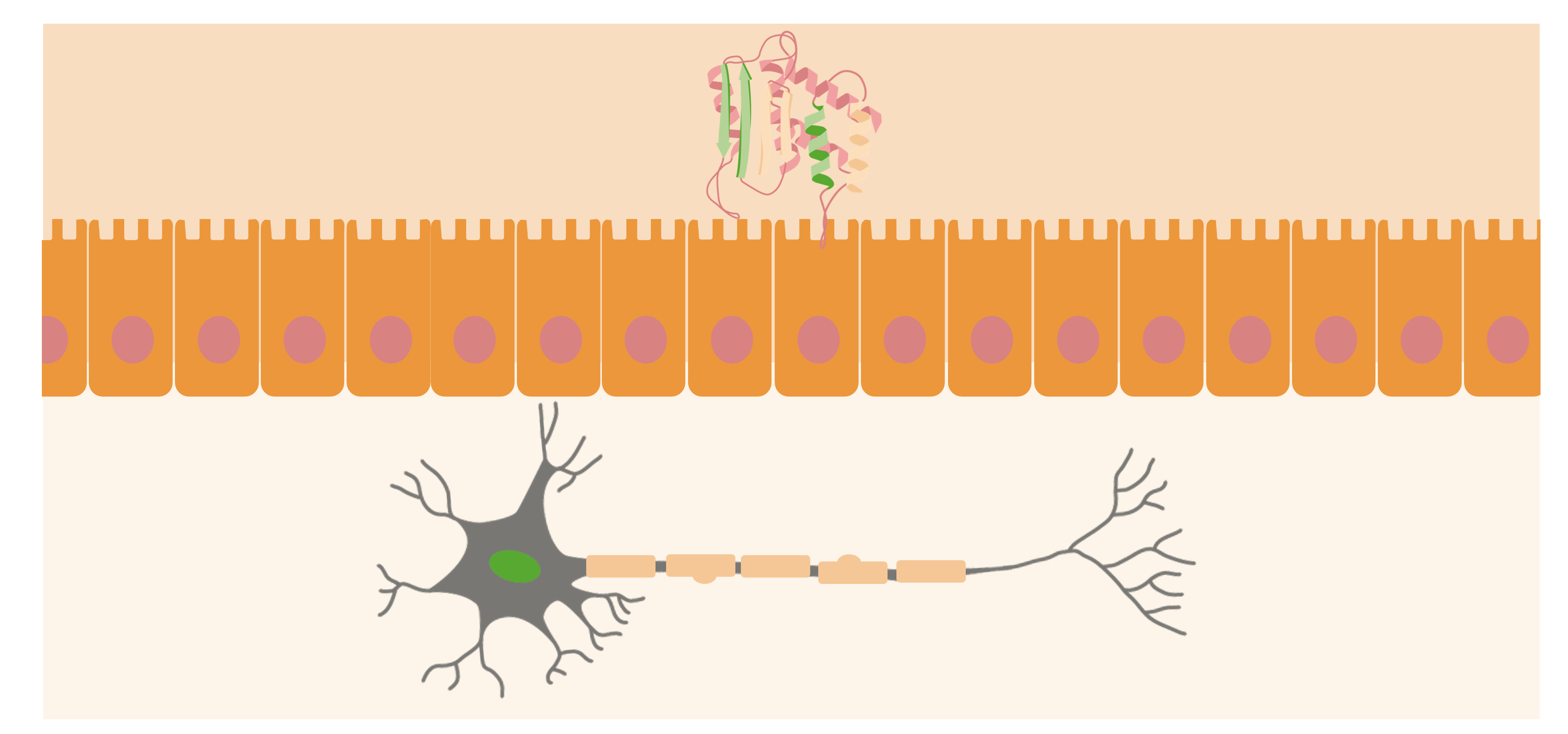
Prions are usually located at the outer surfaces of neurons. When the PrPsc (abnormal protein) is acquired in the organism, it will approach the mucosal surface of the intestine and cause normal prion proteins to convert into pathogenic form.
Image by Lecturio.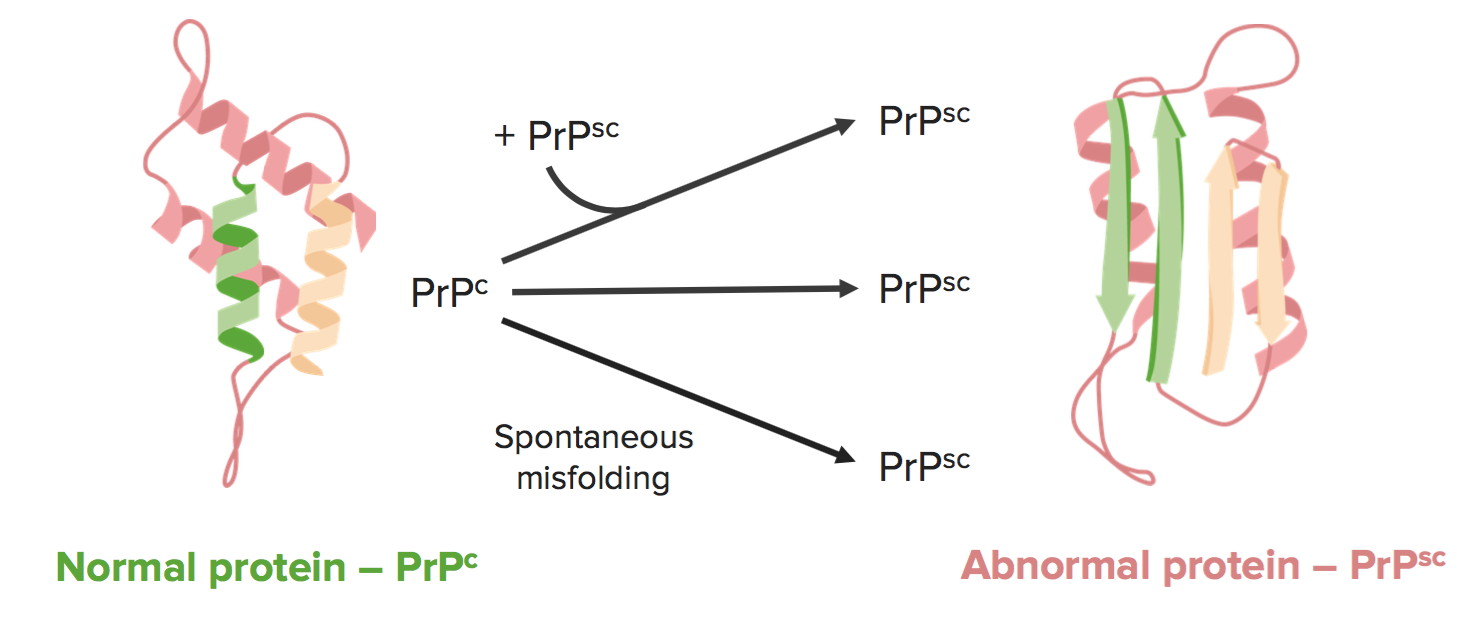
There are 3 primary ways of developing spongiform encephalopathies:
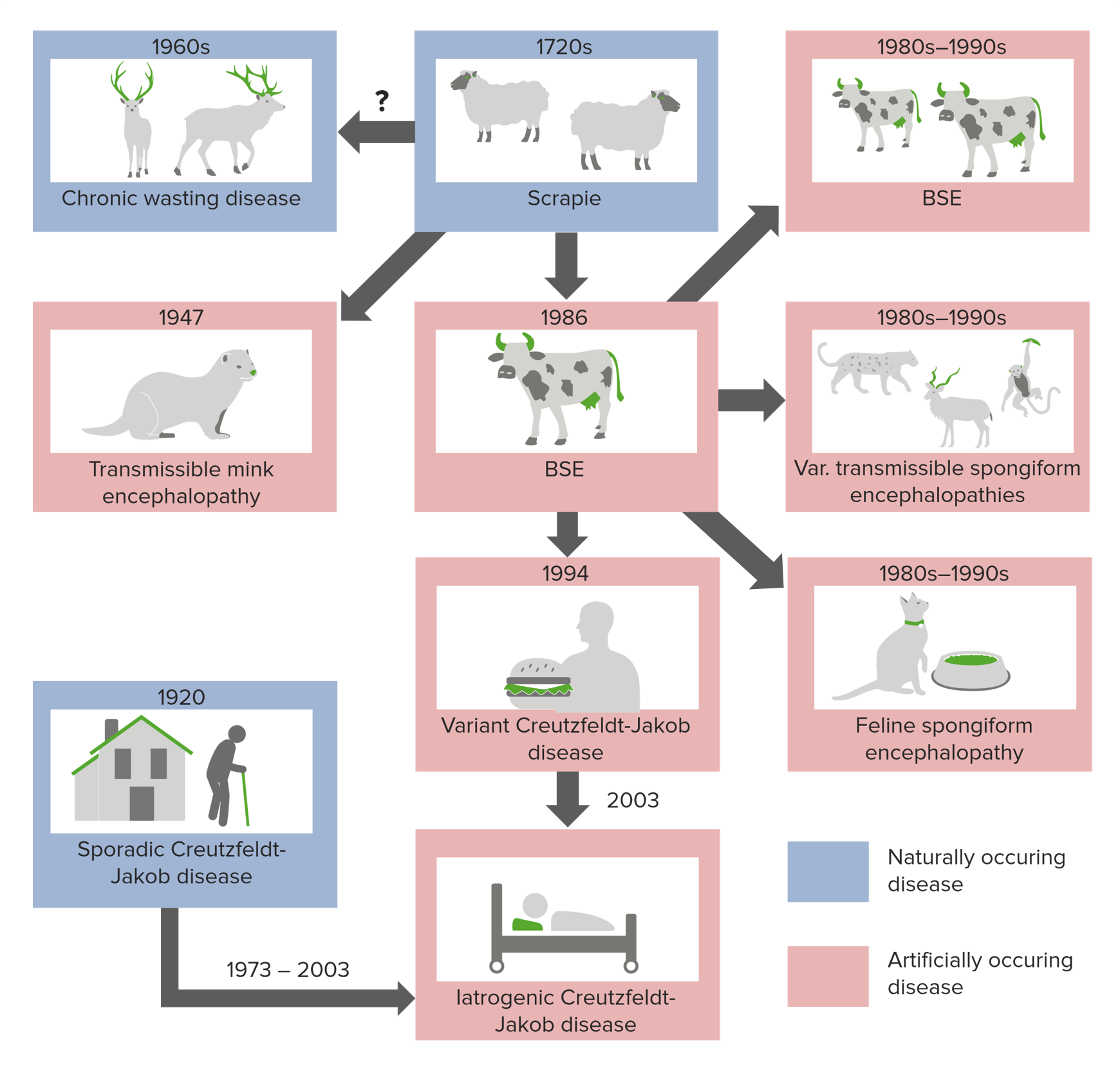
Origins of prion diseases
Image by Lecturio.Transmissible spongiform encephalopathies are associated with extremely long incubation Incubation The amount time between exposure to an infectious agent and becoming symptomatic. Rabies Virus times (20–50 years) and once symptoms appear, the disease rapidly progresses to death.
No curative measures exist and the diseases are universally fatal.
| Symptoms | Diagnosis | |
|---|---|---|
| Creutzfeldt-Jakob disease |
|
|
| Kuru |
|
Unknown; few studies have been performed due to limited cases (mostly isolated to Papua New Guinea in the 1950s) |
| Fatal familial insomnia Insomnia Insomnia is a sleep disorder characterized by difficulty in the initiation, maintenance, and consolidation of sleep, leading to impairment of function. Patients may exhibit symptoms such as difficulty falling asleep, disrupted sleep, trouble going back to sleep, early awakenings, and feeling tired upon waking. Insomnia | In
patients
Patients
Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.
Clinician–Patient Relationship aged 23–73:
|
Unclear; post-mortem brain biopsy Brain biopsy JC Virus and BK Virus |
| Gerstmann-Straussler-Scheinker syndrome | In
patients
Patients
Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.
Clinician–Patient Relationship mid-to-late 40s:
|
Post-mortem brain biopsy Brain biopsy JC Virus and BK Virus |
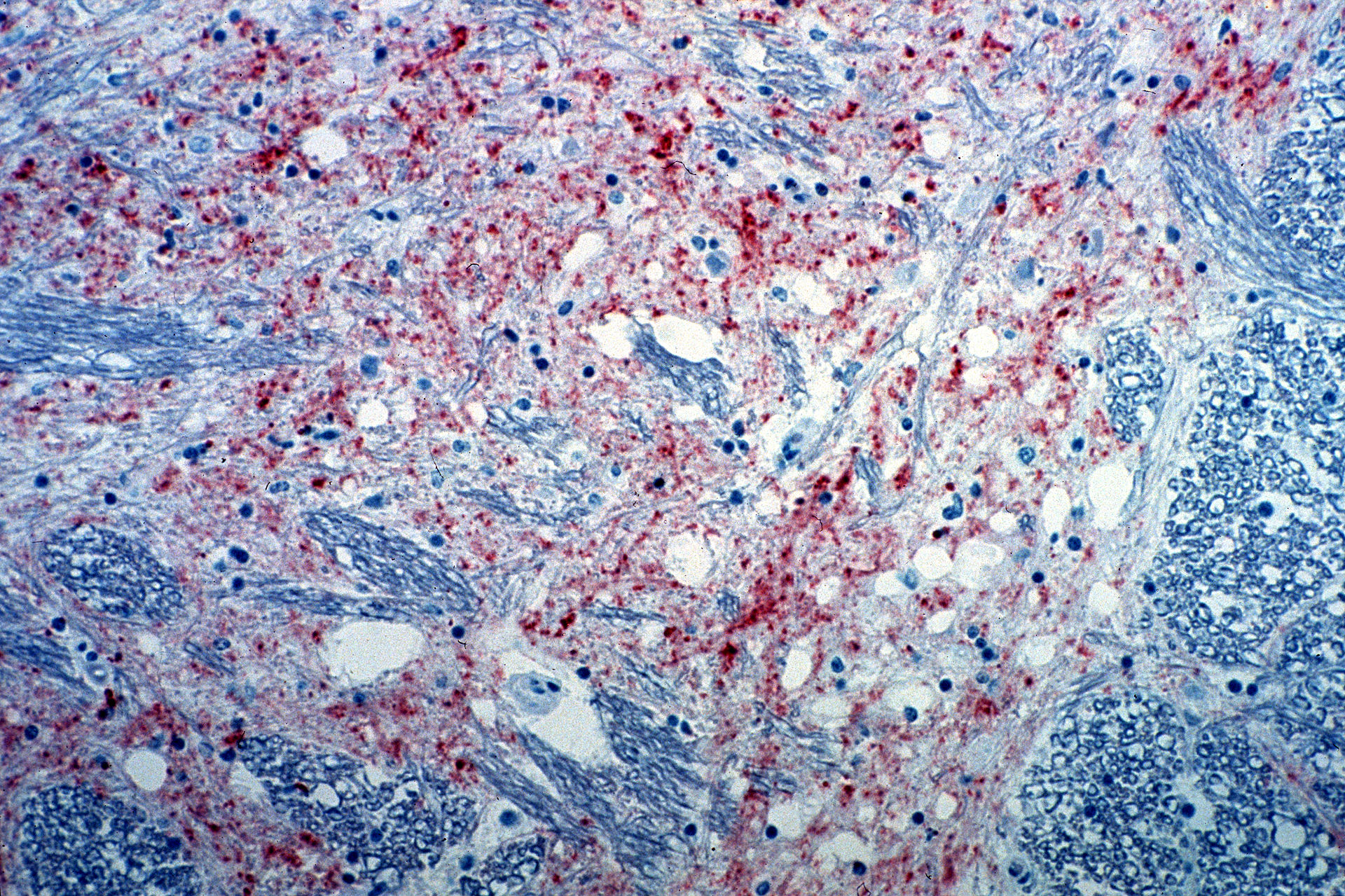
Section of the brain from a cow infected with bovine spongiform encephalopathy. The brown staining indicates the presence of the agent causing the disease.
Image: “Mad_Cow_Disease” by CSIRO. License: CC BY 3.0
Magnified 100 X, and stained with H&E (hematoxylin and eosin) staining technique, this light photomicrograph of brain tissue reveals the presence of prominent spongiotic changes in the cortex and loss of neurons in a case of vCJD.
Image: “Variant Creutzfeldt-Jakob disease (vCJD)” by Sherif Zaki; MD; PhD; Wun-Ju Shieh; MD; PhD; MPH. License: Public DomainDifferential diagnosis includes diseases that are associated with rapidly progressing dementia Dementia Major neurocognitive disorders (NCD), also known as dementia, are a group of diseases characterized by decline in a person’s memory and executive function. These disorders are progressive and persistent diseases that are the leading cause of disability among elderly people worldwide. Major Neurocognitive Disorders, including the following: