


As encefalopatias espongiformes transmissíveis são doenças causadas por priões. Os priões diferem dos vírus por serem pequenos agentes patogénicos infeciosos que não contêm ácido nucleico. As encefalopatias espongiformes incluem a doença de Creutzfeldt-Jakob (DCJ), a variante da doença de Creutzfeldt-Jakob (vDCJ), a Kuru Kuru A prion disease found exclusively among the fore linguistic group natives of the highlands of new guinea. The illness is primarily restricted to adult females and children of both sexes. It is marked by the subacute onset of tremor and ataxia followed by motor weakness and incontinence. Death occurs within 3-6 months of disease onset. The condition is associated with ritual cannibalism, and has become rare since this practice has been discontinued. Pathologic features include a noninflammatory loss of neurons that is most prominent in the cerebellum, glial proliferation, and amyloid plaques. Transmissible Spongiform Encephalopathies, a insónia familiar fatal (IFF) e a síndrome de Gerstmann-Straussler (SGS). Este grupo de doenças apresenta como características comuns a demência, ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia e mioclonia. Infelizmente, estas doenças estão associadas a longos períodos de incubação ( mais MAIS Androgen Insensitivity Syndrome de 20 anos) e, assim que os sintomas ocorrem, progridem rapidamente para a morte.
Last updated: Dec 15, 2025
As encefalopatias espongiformes são extremamente raras.
Pertencem a um grupo de doenças no qual estão incluídas:
A encefalopatia espongiforme mais MAIS Androgen Insensitivity Syndrome comum é a doença de Creutzfeldt-Jakob. Os seus subtipos são:
Os fatores de alto risco são:
As doenças priónicas ocorrem quando uma proteína ⍺ helicoidal normal, conhecida como PrP PRP Raynaud Phenomenonc, é convertida numa proteína β-pregueada anormal, conhecida como PrP PRP Raynaud Phenomenonsc.
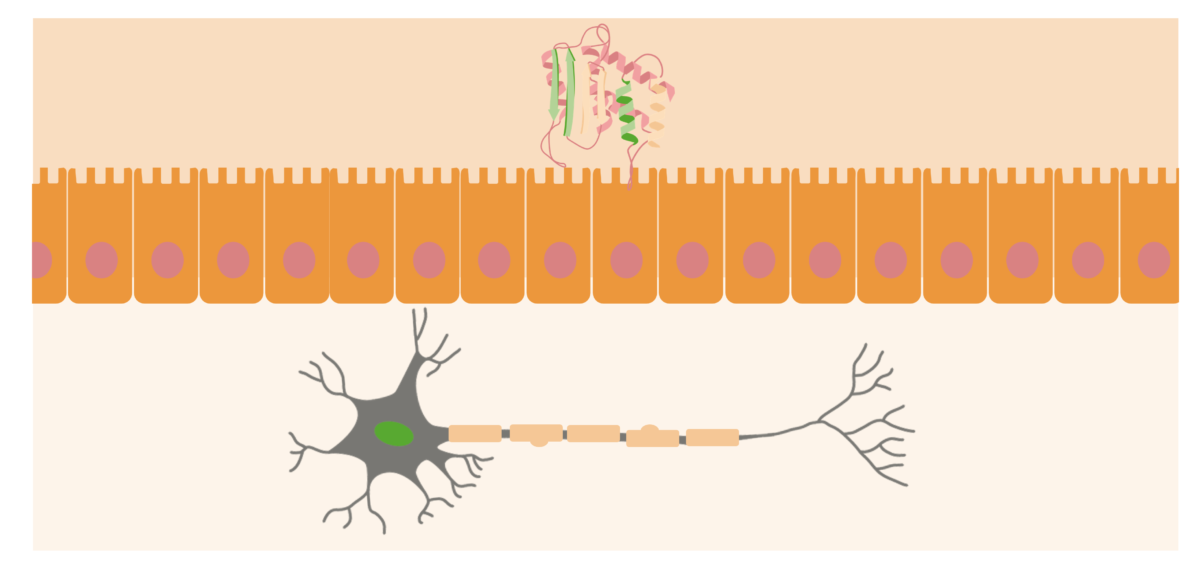
Os priões estão geralmente localizados nas superfícies externas dos neurónios. Quando a PrPSc (proteína anormal) é adquirida no organismo, aproxima-se da superfície mucosa do intestino e leva à conversão das proteínas priónicas normais para a sua forma patogénica.
Imagem por Lecturio.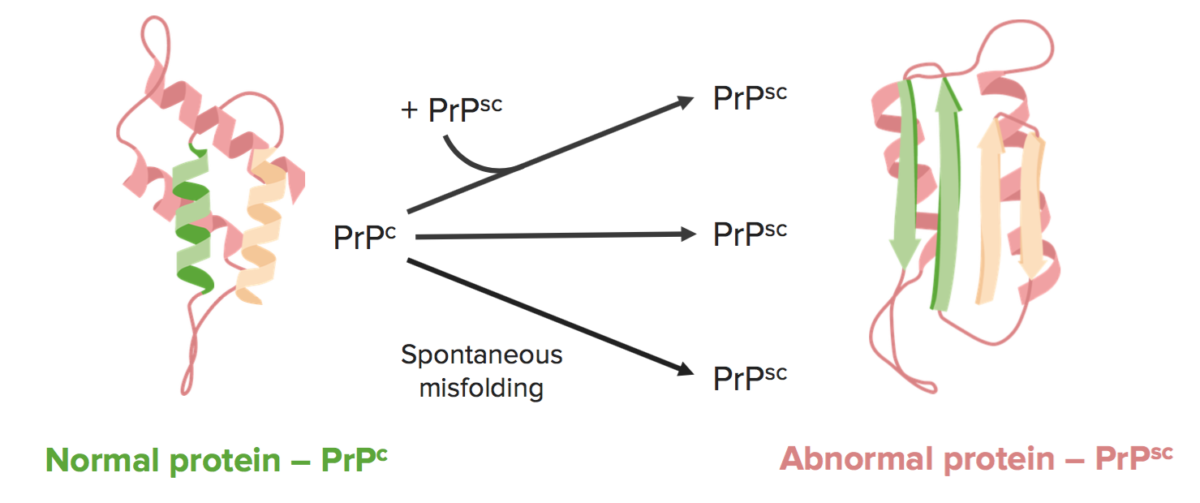
As encefalopatias espongiformes transmissíveis são causadas por dobras incorretas de priões. O prião patogénico, PrPsc, é uma isoforma de conformação de uma proteína hospedeira normal, PrPc
Imagem por Lecturio.Existem 3 formas principais de desenvolver encefalopatia espongiforme:
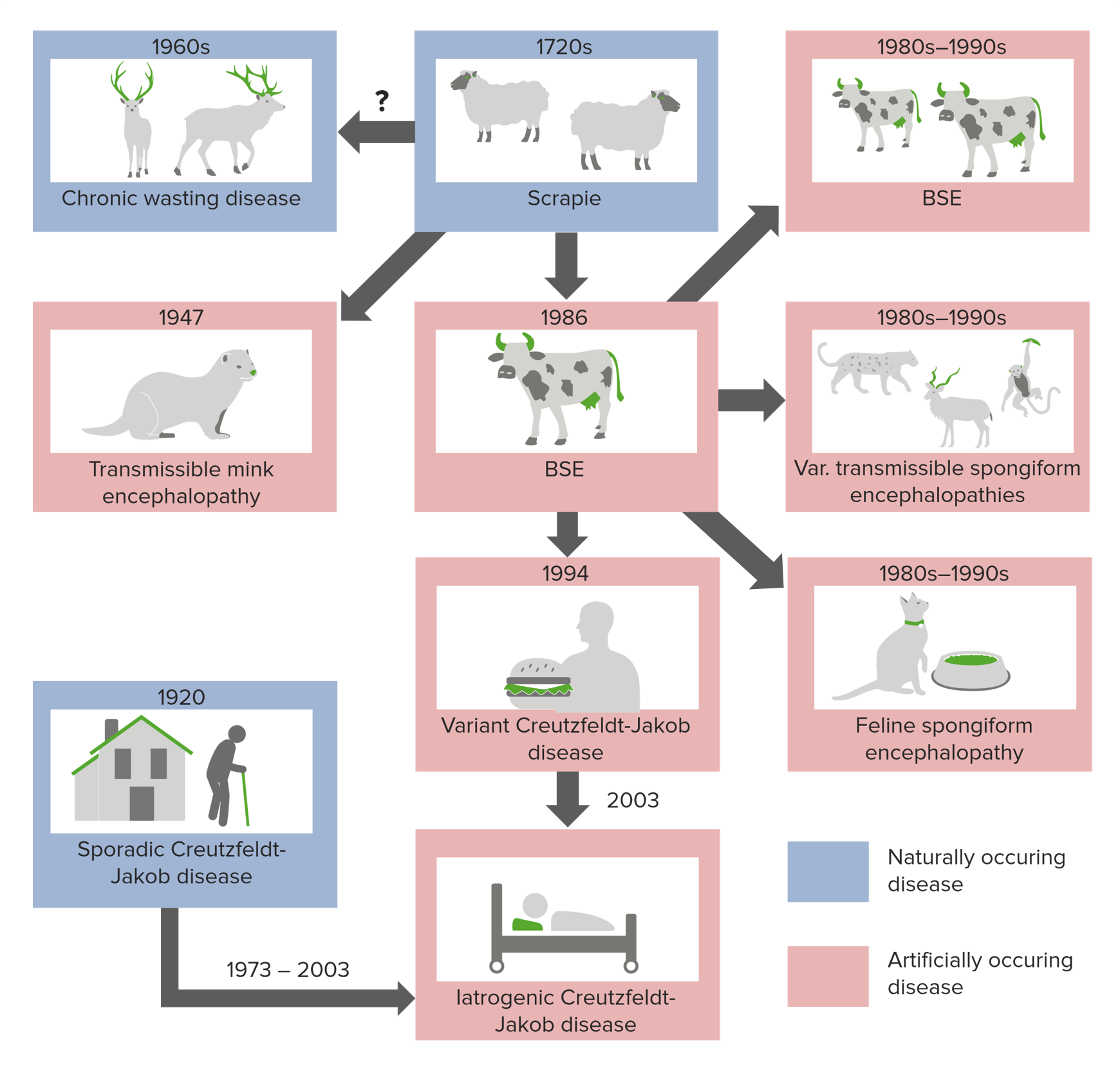
Origem das doenças priónicas
Imagem por Lecturio.As encefalopatias espongiformes transmissíveis estão associadas a tempos de incubação extremamente longos (20 a 50 anos) e, uma vez que os sintomas aparecem, a doença progride rapidamente até à morte.
Não existem medidas curativas e a doença é universalmente fatal.
| Sintomas | Diagnóstico | |
|---|---|---|
| Doença de Creutzfeldt-Jakob |
|
|
| Kuru Kuru A prion disease found exclusively among the fore linguistic group natives of the highlands of new guinea. The illness is primarily restricted to adult females and children of both sexes. It is marked by the subacute onset of tremor and ataxia followed by motor weakness and incontinence. Death occurs within 3-6 months of disease onset. The condition is associated with ritual cannibalism, and has become rare since this practice has been discontinued. Pathologic features include a noninflammatory loss of neurons that is most prominent in the cerebellum, glial proliferation, and amyloid plaques. Transmissible Spongiform Encephalopathies |
|
Desconhecido; poucos estudos realizados devido a casos limitados (principalmente isolados na Papua Nova Guiné na década de 1950) |
| Insónia familiar fatal | Pacientes entre os 23 e 73 anos:
|
Pouco claro; biópsia cerebral post-mortem |
| Síndrome de Gerstmann-Straussler-Scheinker | Pacientes com média de 40 anos:
|
Biópsia cerebral post-mortem |
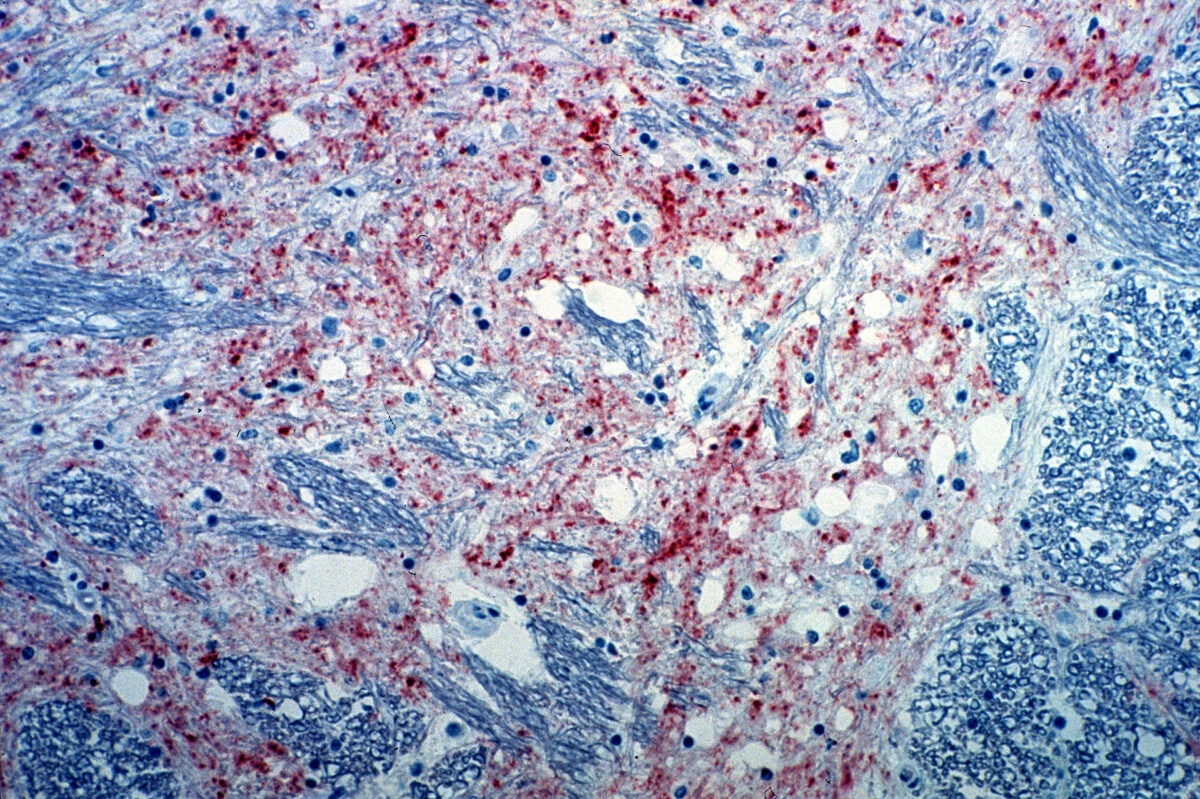
Secção do cérebro de uma vaca infetada com encefalopatia espongiforme bovina. A coloração vermelha indica a presença do agente etiológico da doença.
Imagem : “Mad_Cow_Disease” por CSIRO. Licença: CC BY 3.0
Ampliação 100 X e corada com a técnica de coloração H&E (hematoxilina e eosina), fotomicrografia de tecido cerebral com presença de alterações espongióticas proeminentes no córtex e perda de neurónios, observada num caso de vDCJ.
Imagem : “Variant Creutzfeldt-Jakob disease (vCJD)” por Sherif Zaki; MD; PhD; Wun-Ju Shieh; MD; PhD; MPH. Licença: Domínio PúblicoO diagnóstico diferencial engloba doenças associadas à demência de progressão rápida, incluindo as seguintes:
