Pancreatic neuroendocrine tumors (PanNETs) arise from the endocrine pancreas (islet cells) and represent 1%–2% of primary pancreatic neoplasms; the other > 90% of pancreatic neoplasms are from the exocrine pancreas. The majority of PanNETs are non-functional (50%–75%), while the functional minority may be benign or malignant. Benign or malignant PanNETs include insulinomas, gastrinomas, poorly differentiated pancreatic neuroendocrine carcinomas (NECs), and very rare tumors named glucagonomas, somatostatinomas, and VIPomas, after the hormones they secrete. Diagnosis is made clinically, with laboratory testing for hormone secretion, as well as imaging or upper endoscopy. Management is surgical and may include somatostatin analogs, targeted agents such as everolimus or sunitinib, and peptide-receptor radionuclide therapy (PRRT).
Last updated: Jun 30, 2026
3 of the most common somatic mutations:
Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship may present with metastatic disease in the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy, which is then confirmed with biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma. The diagnostic approach focuses on identifying both the extent of disease spread and the likely primary site of disease.
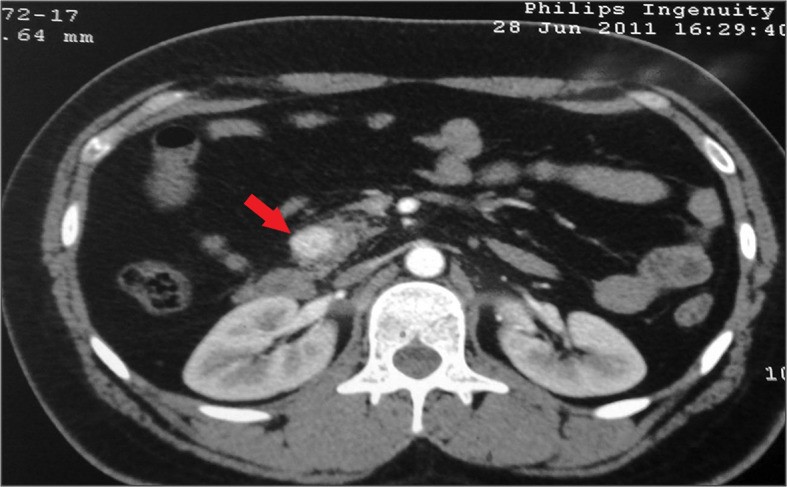
Contrast-enhanced CT (transverse section) showing a solitary insulinoma in the head of the pancreas (arrow)
Image: “Insulinoma presenting as refractory seizure disorder” by Correia P, Panchani R, Ranjan R, Agrawal C. License: CC BY 3.0, edited by Lecturio.In general, management is surgical if the neoplasm is resectable.