


Os tumores neuroendócrinos pancreáticos ( PanNETs PanNETs Pancreatic neuroendocrine tumors (PanNETs) arise from the endocrine pancreas (islet cells) and represent 2%-5% of primary pancreatic neoplasms; the other 95%-98% of pancreatic neoplasms are from the exocrine pancreas. The majority of PanNETs are nonfunctional (50%-75%), while others that are functional may be benign or malignant. Pancreatic Neuroendocrine Tumors (PanNETs), pela sigla em inglês) surgem do pâncreas endócrino (ilhéus) e representam 2%–5% das neoplasias pancreáticas primárias; os outros 95%–98% das neoplasias pancreáticas são do pâncreas exócrino. A maioria dos PanNETs PanNETs Pancreatic neuroendocrine tumors (PanNETs) arise from the endocrine pancreas (islet cells) and represent 2%-5% of primary pancreatic neoplasms; the other 95%-98% of pancreatic neoplasms are from the exocrine pancreas. The majority of PanNETs are nonfunctional (50%-75%), while others that are functional may be benign or malignant. Pancreatic Neuroendocrine Tumors (PanNETs) são não funcionantes (50%–75%), enquanto que os funcionantes podem ser benignos ou malignos. Os PanNETs PanNETs Pancreatic neuroendocrine tumors (PanNETs) arise from the endocrine pancreas (islet cells) and represent 2%-5% of primary pancreatic neoplasms; the other 95%-98% of pancreatic neoplasms are from the exocrine pancreas. The majority of PanNETs are nonfunctional (50%-75%), while others that are functional may be benign or malignant. Pancreatic Neuroendocrine Tumors (PanNETs) benignos ou malignos incluem os insulinomas Insulinomas An insulinoma is a type of functional neuroendocrine tumor that manifests with hypoglycemia due to autologous secretion of insulin. It more commonly presents as a solitary benign tumor but can sometimes be associated with MEN type 1 (MEN1). Patients present with fasting hypoglycemia, which may manifest as episodes of diaphoresis, palpitations, tremor, and confusion. Insulinomas, gastrinomas, carcinomas neuroendócrinos pancreáticos malignos e tumores muito raros denominados glucagonomas, somatostatinomas e VIPomas, devido às hormonas que secretam. O diagnóstico é feito clinicamente, com exames laboratoriais para averiguação da secreção hormonal, bem como exames de imagem ou endoscopia digestiva alta. O tratamento é cirúrgico ou com terapêuticas molecularmente dirigidas mais MAIS Androgen Insensitivity Syndrome recentes.
Last updated: Jun 30, 2026
São 3 as mutações somáticas mais MAIS Androgen Insensitivity Syndrome comuns:
Os doentes podem apresentar doença metastática hepática, confirmada através da realização de biópsia. A abordagem diagnóstica concentra-se na identificação tanto da extensão da disseminação da doença quanto do provável local primário da doença.
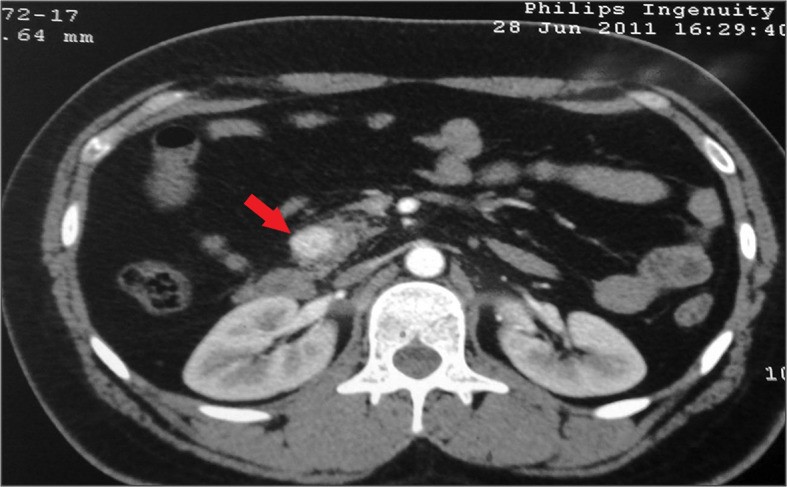
TC com contraste (corte transversal) que evidencia um insulinoma solitário na cabeça do pâncreas (seta)
Imagem : “Insulinoma presenting as refractory seizure disorder” por Correia P, Panchani R, Ranjan R, Agrawal C. Licença: CC BY 3.0, editado por Lecturio.Em geral, o tratamento é cirúrgico se a neoplasia for ressecável.
