Wilms tumor Tumor Inflammation is a malignancy Malignancy Hemothorax caused by proliferation of metanephric blastema in the kidneys Kidneys The kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine. Kidneys: Anatomy and is the most common renal malignancy Malignancy Hemothorax in children. Wilms tumor Tumor Inflammation usually arises sporadically, but it can also occur as a result of a specific congenital anomaly like WAGR (Wilms tumor Tumor Inflammation, aniridia, genitourinary abnormalities, and mental retardation) syndrome, Denys–Drash syndrome Denys–Drash Syndrome Nephrotic Syndrome in Children, or Beckwith–Wiedemann syndrome. Wilms tumor Tumor Inflammation commonly presents as a firm, nontender, smooth mass Mass Three-dimensional lesion that occupies a space within the breast Imaging of the Breast that does not cross the midline. Wilms tumor Tumor Inflammation can also rarely present with abdominal pain Abdominal Pain Acute Abdomen, hematuria Hematuria Presence of blood in the urine. Renal Cell Carcinoma, and/or hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension. The malignancy Malignancy Hemothorax is diagnosed with abdominal ultrasonography and histopathologic studies (from biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma or resection). Wilms tumor Tumor Inflammation is treated with multimodal therapy (surgery, chemotherapy Chemotherapy Osteosarcoma, radiation Radiation Emission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles). Osteosarcoma). Influenced by patient age, molecular markers, and pathologic findings, prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas is favorable overall, with 5-year survival rates approaching 90%.
Last updated: Dec 15, 2025
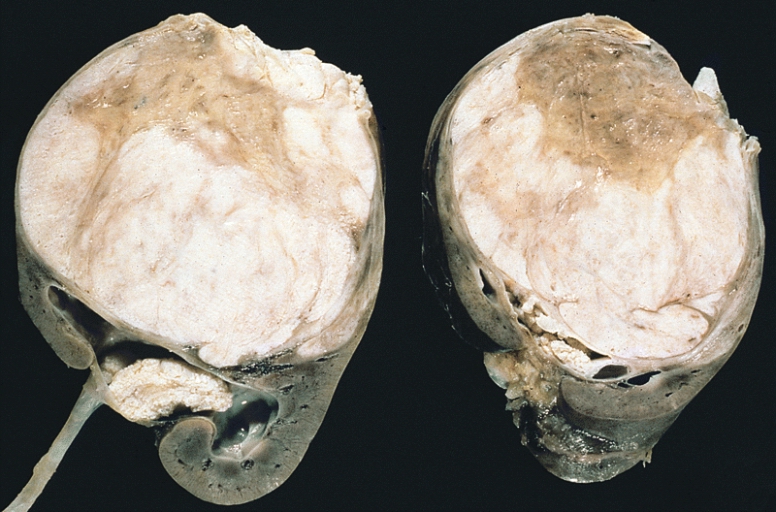
Wilms tumor:
Note the prominent septa subdividing the sectioned surface and the protrusion of tumor into the renal pelvis, resembling botryoid rhabdomyosarcoma.
Serial abdominal ultrasonography for those at risk for Wilms tumor Tumor Inflammation:
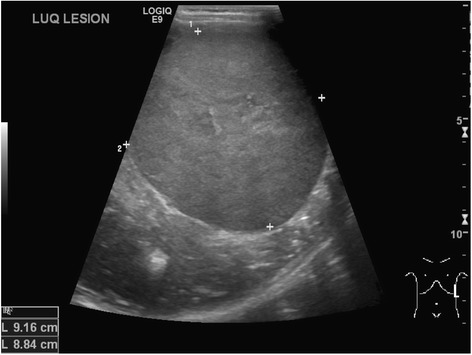
Abdominal ultrasound revealing left-sided Wilms tumor in 4-year-old boy
Image: “Neuroblastoma and nephroblastoma: a radiological review” by Dumba M, Jawad N, McHugh K. License: CC BY 4.0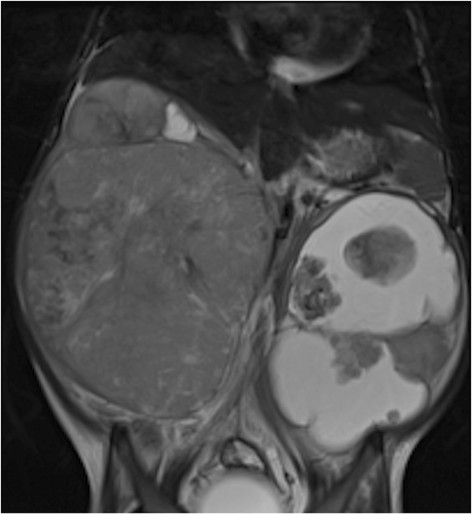
MRI of Wilms tumor: 4-year-old girl with bilateral Wilms’ tumor, more cystic on the left
Image: “Figure 11” by Dumba, M. et al. License: CC BY 4.0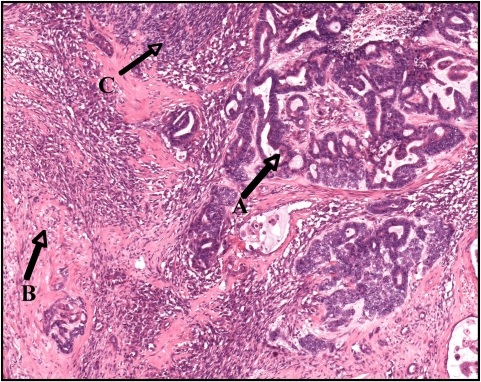
Wilms tumor exhibiting an epithelial component (A), stromal component (B), and small areas of blastema (C)
Image: “Wilms tumor in a 37-year-old” by Thevendran G, Farne HA, Kaisary AV. License: CC BY 2.0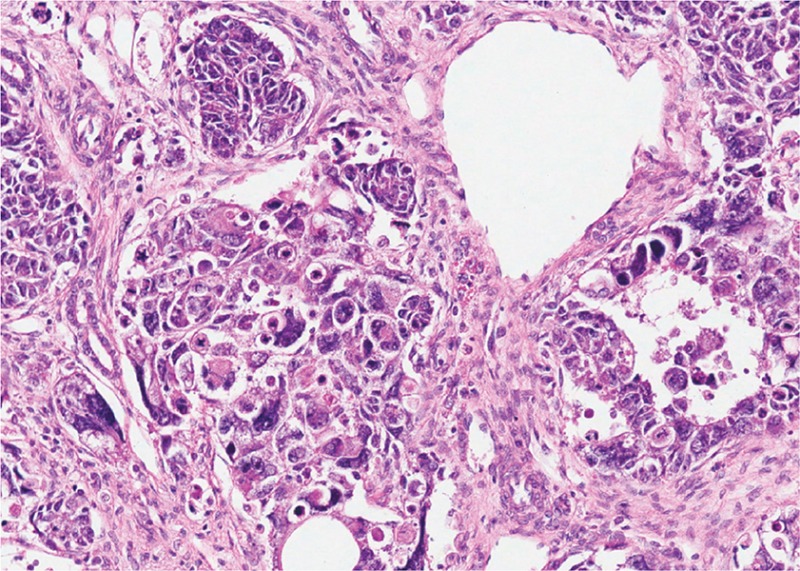
Histopathologic examination from biopsy of Wilms tumor with anaplasia
Image: “Wilms tumour with anaplasia” by van den Heuvel-Eibrink, MM. License: CC BY 4.0From National Wilms Tumor Tumor Inflammation Study (NWTS)/Children’s Oncology Group (COG):
Recommendations based on the NWTS/COG trials: