


O tumor Tumor Inflammation de Wilms é uma doença maligna causada pela proliferação de blastema metanéfrico nos rins e é a neoplasia renal mais MAIS Androgen Insensitivity Syndrome comum em crianças. O tumor Tumor Inflammation de Wilms surge geralmente de forma esporádica, mas também pode ocorrer como resultado de uma anomalia congénita específica como a síndrome WAGR ( tumor Tumor Inflammation de Wilms, aniridia Aniridia A congenital abnormality in which there is only a rudimentary iris. This is due to the failure of the optic cup to grow. Aniridia also occurs in a hereditary form, usually autosomal dominant. Wilms Tumor, anomalias geniturinárias e défice cognitivo), síndrome de Denys-Drash ou síndrome de Beckwith-Wiedemann. O tumor Tumor Inflammation de Wilms apresenta-se frequentemente como uma massa firme, sem desconforto abdominal, lisa, que não cruza a linha média. O tumor Tumor Inflammation de Wilms também se pode apresentar, raramente, com dor abdominal, hematúria e / ou hipertensão. A malignidade é diagnosticada com ecografia abdominal e estudos histopatológicos (por biópsia ou resseção). O tumor Tumor Inflammation de Wilms é tratado com terapia multimodal (cirurgia, quimioterapia, radiação). Influenciado pela idade do doente, marcadores moleculares e achados patológicos, o prognóstico é globalmente favorável, com taxas de sobrevida aos 5 anos de aproximadamente 90%.
Last updated: Dec 15, 2025
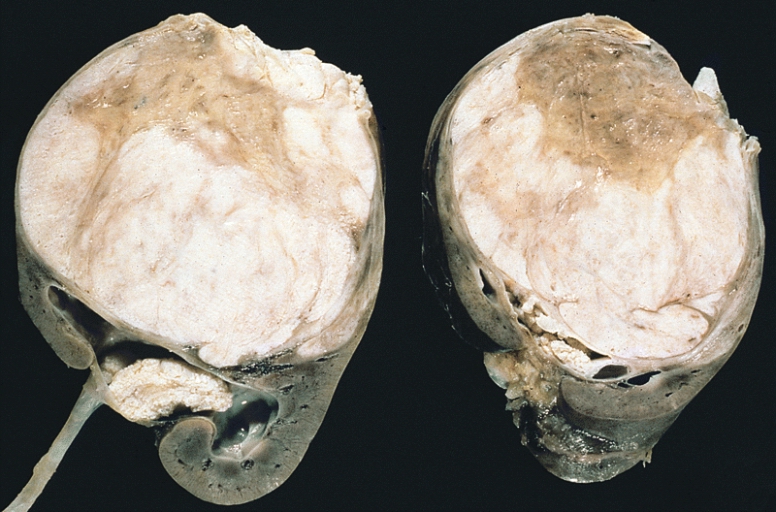
Tumor de Wilms:
Observar os septos proeminentes que subdividem a superfície seccionada e a protrusão do tumor na pelve renal, que assemelha um rabdomiossarcoma botrióide.
Ecografia abdominal seriada para aqueles em risco de tumor Tumor Inflammation de Wilms:
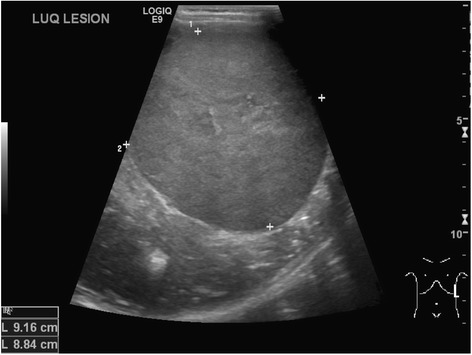
Ecografia abdominal revela tumor de Wilms do lado esquerdo num menino de 4 anos
Imagem: “Neuroblastoma and nephroblastoma: a radiological review” por Dumba M, Jawad N, McHugh K. Licença: CC BY 4.0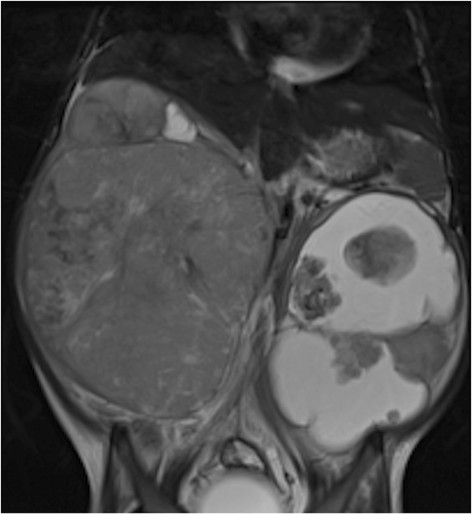
Ressonância magnética de tumor de Wilms: menina de 4 anos com tumor de Wilms bilateral, mais quístico à esquerda
Imagem: “Figure 11” por Dumba, M. et al. Licença: CC BY 4.0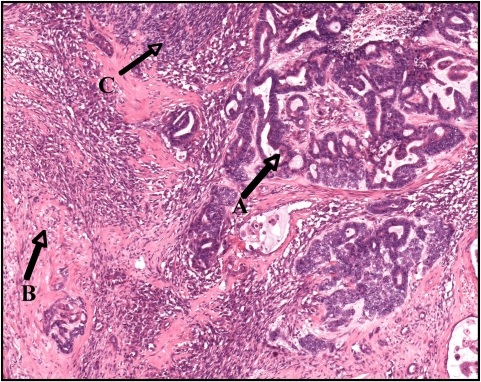
Tumor de Wilms com componente epitelial (A), componente estromal (B), e pequenas áreas de blastema (C)
Imagem: “Wilms tumor in a 37-year-old” por Thevendran G, Farne HA, Kaisary AV. Licença: CC BY 2.0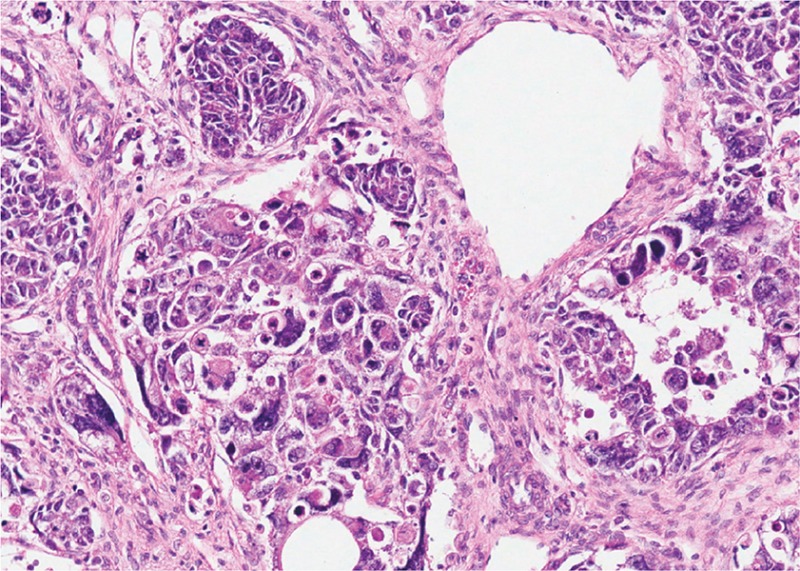
Exame histopatológico da biópsia de tumor de Wilms com anaplasia
Imagem: “Wilms tumour with anaplasia” por van den Heuvel-Eibrink, MM. Licença: CC BY 4.0Do National Wilms Tumor Tumor Inflammation Study (NWTS) / Children’s Oncology Group (COG):
Recomendações baseadas nos ensaios NWTS / COG:
