Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts Cysts Any fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues. Fibrocystic Change in the kidneys Kidneys The kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine. Kidneys: Anatomy. The 2 main types of PKD are autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance polycystic kidney disease (ADPKD), which is often diagnosed in adulthood, and autosomal recessive polycystic kidney disease Autosomal Recessive Polycystic Kidney Disease Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts in the kidneys. Autosomal recessive polycystic kidney disease is primarily characterized by cystic dilatations of the renal collecting ducts and intrahepatic bile duct dilatation with hepatic fibrosis. Autosomal Recessive Polycystic Kidney Disease (ARPKD) ( ARPKD ARPKD Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts in the kidneys. Autosomal recessive polycystic kidney disease is primarily characterized by cystic dilatations of the renal collecting ducts and intrahepatic bile duct dilatation with hepatic fibrosis. Autosomal Recessive Polycystic Kidney Disease (ARPKD)), which is often diagnosed antenatally or shortly after birth. ADPKD patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship commonly present with hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, hematuria Hematuria Presence of blood in the urine. Renal Cell Carcinoma, and flank pain Flank pain Pain emanating from below the ribs and above the ilium. Renal Cell Carcinoma. Extrarenal manifestations include intracerebral aneurysm Aneurysm An aneurysm is a bulging, weakened area of a blood vessel that causes an abnormal widening of its diameter > 1.5 times the size of the native vessel. Aneurysms occur more often in arteries than in veins and are at risk of dissection and rupture, which can be life-threatening. Thoracic Aortic Aneurysms, hepatic and pancreatic cysts Cysts Any fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues. Fibrocystic Change, and cardiac valvular abnormalities. Diagnosis is by history, physical exam, and ultrasonography. Management requires a multidisciplinary approach and many patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship require renal replacement therapy. The end goal is to slow the progression of renal disease by controlling hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, proteinuria Proteinuria The presence of proteins in the urine, an indicator of kidney diseases. Nephrotic Syndrome in Children, and symptoms. ADPKD prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas is dependent on a variety of factors. A cerebral aneurysm Cerebral aneurysm Brain aneurysms, also known as intracranial or cerebral aneurysms, are dilations of the arteries along points of weakness in the brain. The majority of the aneurysms are berry (saccular) in nature and located within the anterior circulation of the circle of Willis. Brain Aneurysms is a complication associated with a particularly poor prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas.
Last updated: Dec 15, 2025
Polycystic kidney disease (PKD) affects about 500,000 people in the United States. One of the 2 main types of PKD is autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance polycystic kidney disease (ADPKD).
The age of symptom onset is variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables, but typically in adulthood. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with PKD1 mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations present with symptoms earlier than patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with PKD2.
Diagnosis is made with the combination of positive family history Family History Adult Health Maintenance and the presence of multiple, bilateral renal cysts Renal Cysts Imaging of the Urinary System.

Ultrasound of autosomal dominant polycystic kidney disease (ADPKD):
sagittal gray-scale ultrasound scan of both kidneys shows numerous cystic, space-occupying lesions with marked internal echogenic debris and thick septa, representing the numerous infected cysts
Goals:
Treatment measures:
For end-stage renal disease (ESRD):
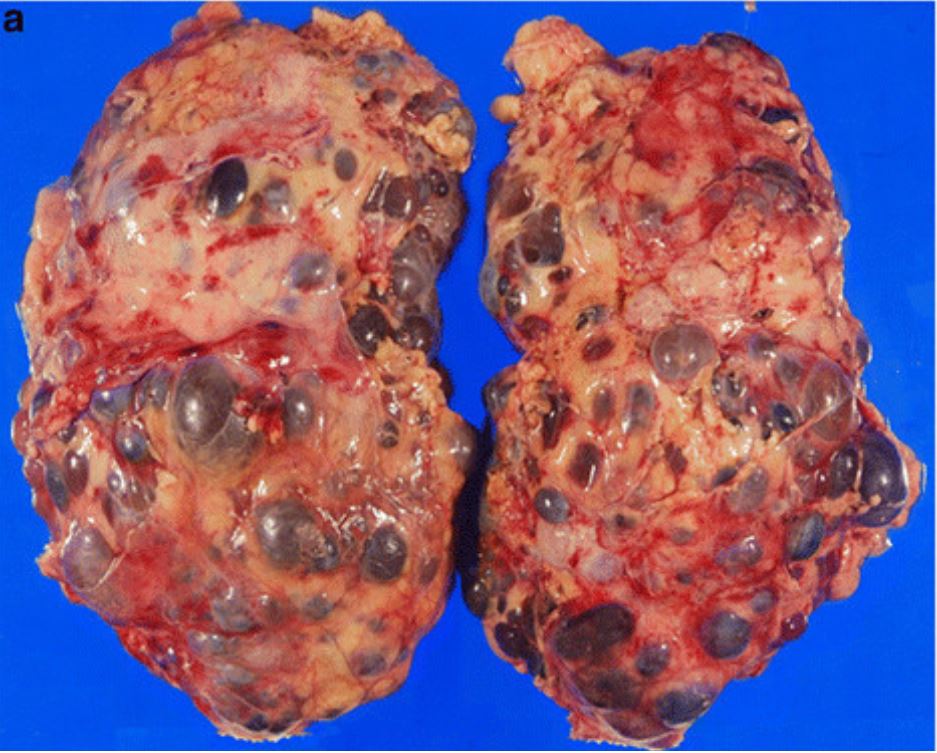
Gross pathology from nephrectomy in a patient with autosomal dominant polycystic kidney disease (ADPKD)
Image: “Incidental renal cell carcinoma presenting in a renal transplant recipient with autosomal dominant polycystic kidney disease” by Misumi T, Ide K, Onoe T, Banshodani M, Tazawa H, Teraoka Y, Hotta R, Yamashita M, Tashiro H, Ohdan H. License: CC BY 2.0, cropped by Lecturio.The 2 main types of polycystic kidney disease are ADPKD and autosomal recessive polycystic kidney disease Autosomal Recessive Polycystic Kidney Disease Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts in the kidneys. Autosomal recessive polycystic kidney disease is primarily characterized by cystic dilatations of the renal collecting ducts and intrahepatic bile duct dilatation with hepatic fibrosis. Autosomal Recessive Polycystic Kidney Disease (ARPKD) ( ARPKD ARPKD Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts in the kidneys. Autosomal recessive polycystic kidney disease is primarily characterized by cystic dilatations of the renal collecting ducts and intrahepatic bile duct dilatation with hepatic fibrosis. Autosomal Recessive Polycystic Kidney Disease (ARPKD)).
| ADPKD | ARPKD ARPKD Polycystic kidney disease (PKD) is an inherited genetic disorder leading to the development of numerous fluid-filled cysts in the kidneys. Autosomal recessive polycystic kidney disease is primarily characterized by cystic dilatations of the renal collecting ducts and intrahepatic bile duct dilatation with hepatic fibrosis. Autosomal Recessive Polycystic Kidney Disease (ARPKD) | |
|---|---|---|
| Inheritance | Autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance | Autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance |
| Genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure involved | PKD1, PKD2 | PKHD1 PKHD1 Autosomal Recessive Polycystic Kidney Disease (ARPKD) |
| Associated proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis | Polycystin-1, polycystin-2 | Fibrocystin |
| Age of presentation | Adulthood | Antenatal, neonatal, infant |
| Clinical features |
|
|
| Gross and pathologic morphology |
|
|
| Ultrasound findings |
Multiple
cysts
Cysts
Any fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues.
Fibrocystic Change (based on age):
|
|