Chronic granulomatous disease Granulomatous disease A defect of leukocyte function in which phagocytic cells ingest but fail to digest bacteria, resulting in recurring bacterial infections with granuloma formation. When chronic granulomatous disease is caused by mutations in the cybb gene, the condition is inherited in an X-linked recessive pattern. When chronic granulomatous disease is caused by cyba, ncf1, ncf2, or ncf4 gene mutations, the condition is inherited in an autosomal recessive pattern. Common Variable Immunodeficiency (CVID) (CGD), as the name implies, is a chronic disorder that is characterized by granuloma formation. This disorder is a consequence of defective phagocytic cells that are unable to produce bactericidal Bactericidal Penicillins superoxide because of a defect in nicotinamide adenine dinucleotide Nicotinamide adenine dinucleotide A coenzyme composed of ribosylnicotinamide 5'-diphosphate coupled to adenosine 5'-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway phosphate Phosphate Inorganic salts of phosphoric acid. Electrolytes ( NADPH NADPH Nicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5'-phosphate (nmn) coupled by pyrophosphate linkage to the 5'-phosphate adenosine 2. Pentose Phosphate Pathway), the oxidase Oxidase Neisseria responsible for the respiratory burst Respiratory burst A large increase in oxygen uptake by neutrophils and most types of tissue macrophages through activation of an NADPH-cytochrome b-dependent oxidase that reduces oxygen to a superoxide. Individuals with an inherited defect in which the oxidase that reduces oxygen to superoxide is decreased or absent often die as a result of recurrent bacterial infections. Leukocyte Adhesion Deficiency Type 1 in phagocytic leukocytes Leukocytes White blood cells. These include granular leukocytes (basophils; eosinophils; and neutrophils) as well as non-granular leukocytes (lymphocytes and monocytes). White Myeloid Cells: Histology. The diagnosis is made by testing neutrophil function for superoxide production. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with CGD are at increased risk of life-threatening infections with fungi Fungi A kingdom of eukaryotic, heterotrophic organisms that live parasitically as saprobes, including mushrooms; yeasts; smuts, molds, etc. They reproduce either sexually or asexually, and have life cycles that range from simple to complex. Filamentous fungi, commonly known as molds, refer to those that grow as multicellular colonies. Mycology and catalase-positive bacteria Bacteria Bacteria are prokaryotic single-celled microorganisms that are metabolically active and divide by binary fission. Some of these organisms play a significant role in the pathogenesis of diseases. Bacteriology. Inflammatory complications such as CGD colitis Colitis Inflammation of the colon section of the large intestine, usually with symptoms such as diarrhea (often with blood and mucus), abdominal pain, and fever. Pseudomembranous Colitis are also possible. The introduction of antimicrobial prophylaxis Prophylaxis Cephalosporins and the use of azole antifungals has increased the overall life expectancy Life expectancy Based on known statistical data, the number of years which any person of a given age may reasonably expected to live. Population Pyramids of these patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship.
Last updated: Dec 15, 2025
Chronic granulomatous disease Granulomatous disease A defect of leukocyte function in which phagocytic cells ingest but fail to digest bacteria, resulting in recurring bacterial infections with granuloma formation. When chronic granulomatous disease is caused by mutations in the cybb gene, the condition is inherited in an X-linked recessive pattern. When chronic granulomatous disease is caused by cyba, ncf1, ncf2, or ncf4 gene mutations, the condition is inherited in an autosomal recessive pattern. Common Variable Immunodeficiency (CVID) (CGD) is caused by mutations in the genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure encoding for proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis that make up the nicotinamide adenine dinucleotide Nicotinamide adenine dinucleotide A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway phosphate Phosphate Inorganic salts of phosphoric acid. Electrolytes ( NADPH NADPH Nicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2. Pentose Phosphate Pathway) oxidase Oxidase Neisseria:
The NADPH-oxidase enzyme is impaired in phagocytic cell → ↓ production of superoxide anion O2–, which affects other oxidants. This enzyme is responsible for the inability to fight infections.
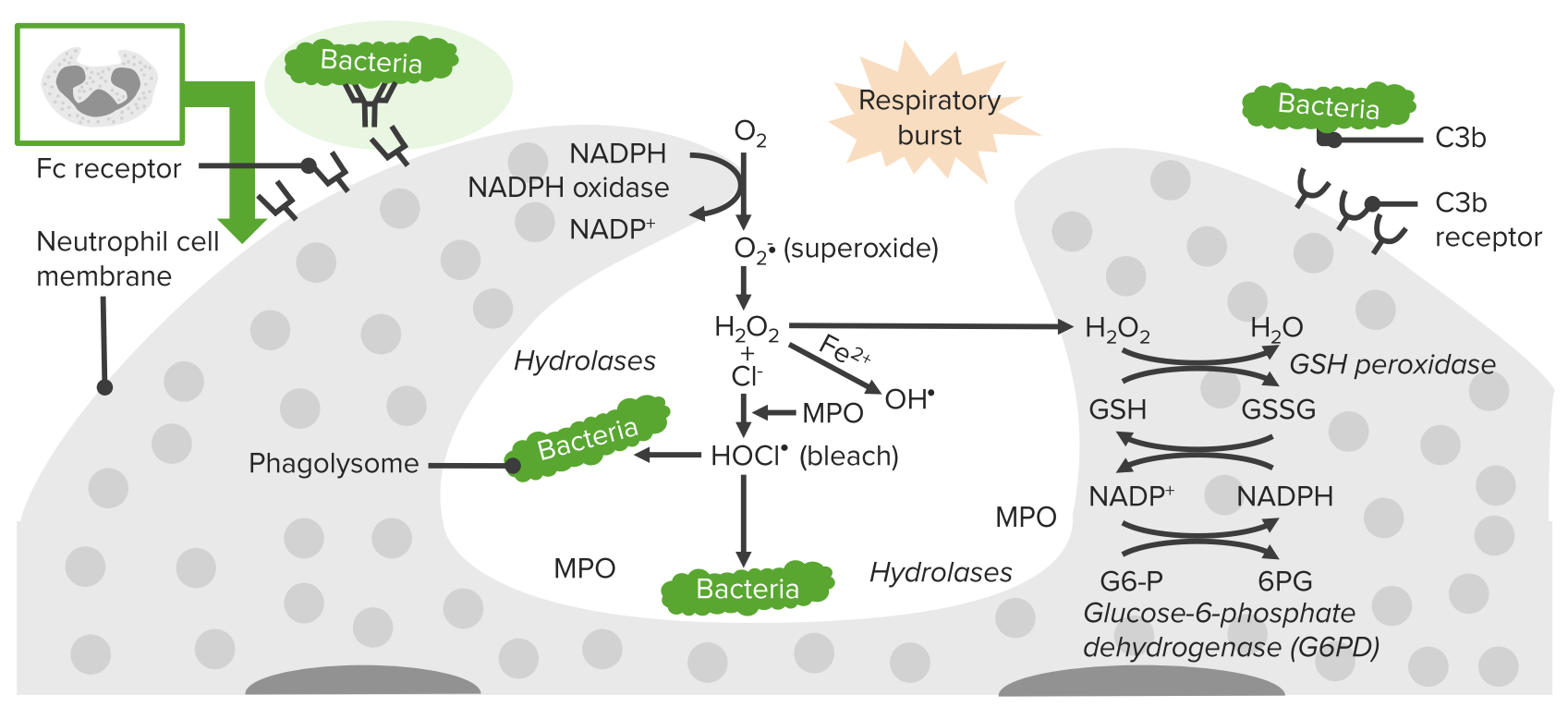
Process of phagocytosis in neutrophils:
A deficiency in NADPH oxidase results in an inability to produce superoxide, which diminishes the cell’s ability to destroy catalase-positive microorganisms.
Most CGD patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship are < 5 years of age, and the severity is variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables (depending on the genetic defect Genetic Defect Ion Channel Myopathy). Clinical presentation can include:
Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship may experience repeated infections caused by bacterial and fungal pathogens.
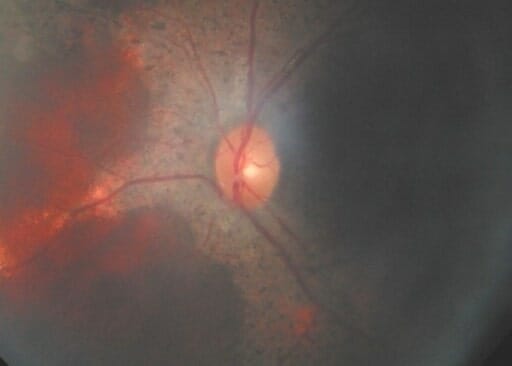
The fundus of an individual with chronic granulomatous disease (CGD) showing a retinal mass
Image: “F3: Brother’s fundus” by Mansour A. M. et al. License: CC BY 2.0Neutrophil function tests should be done:
Molecular genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies and genotyping Genotyping Methods used to determine individuals’ specific alleles or snps (single nucleotide polymorphisms). Polymerase Chain Reaction (PCR):
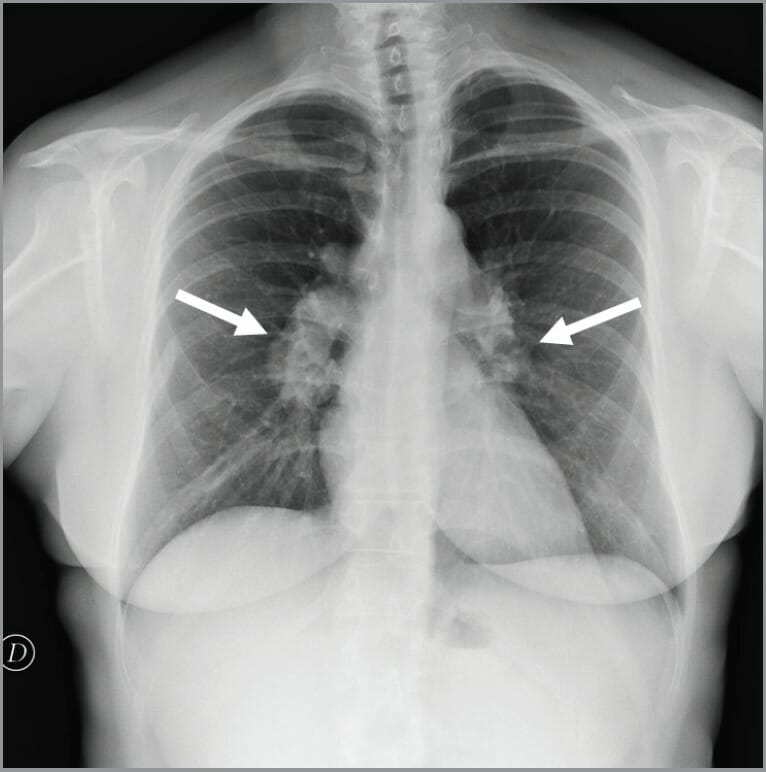
Chest X-ray showing bilateral hilar lymphadenopathy in an individual with chronic granulomatous disease (CGD)
Image: “Chest x-ray showed bilateral hilar lymphadenopathies (arrows) suggestive for chronic granulomatous disease” by Conte G. et al. License: CC BY 3.0