


A doença granulomatosa crónica (DGC), como o nome indica, é uma doença crónica caracterizada pela formação de granulomas Granulomas A relatively small nodular inflammatory lesion containing grouped mononuclear phagocytes, caused by infectious and noninfectious agents. Sarcoidosis. Esta doença é uma consequência da incapacidade de células fagocíticas defeituosas em produzir superóxido bactericida devido a um defeito na nicotinamida adenina dinucleotídeo fosfato ( NADPH NADPH Nicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5'-phosphate (nmn) coupled by pyrophosphate linkage to the 5'-phosphate adenosine 2. Pentose Phosphate Pathway, pela sigla em inglês), a oxidase Oxidase Neisseria responsável pelo aumento de uptake respiratório de O2 nos leucócitos fagocitários. O diagnóstico é feito pela testagem da função dos neutrófilos para a produção de superóxido. Doentes com DGC estão em maior risco de infeções com risco de vida por fungos e bactérias catalase-positivas. Também são possíveis complicações inflamatórias como colite CGD CGD Chronic granulomatous disease (CGD), as the name implies, is a chronic disorder that is characterized by granuloma formation. This disorder is a consequence of defective phagocytic cells that are unable to produce bactericidal superoxide because of a defect in nicotinamide adenine dinucleotide phosphate (NADPH), the oxidase responsible for the respiratory burst in phagocytic leukocytes. Chronic Granulomatous Disease. A introdução da profilaxia antimicrobiana e o uso de antifúngicos azólicos aumentam a esperança de vida destes doentes.
Last updated: Dec 15, 2025
A doença granulomatosa crónica (DGC) é causada por mutações nos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codificam os constituintes da nicotinamida adenina dinucleotídeo fosfato ( NADPH NADPH Nicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2. Pentose Phosphate Pathway, pela sigla em inglês) oxidase Oxidase Neisseria:
A enzima NADPH-oxidase está afetada na célula fagocitária → ↓ produção do anião superóxido O 2– , que afeta outros oxidantes. Esta enzima é responsável pela incapacidade de combater infeções.
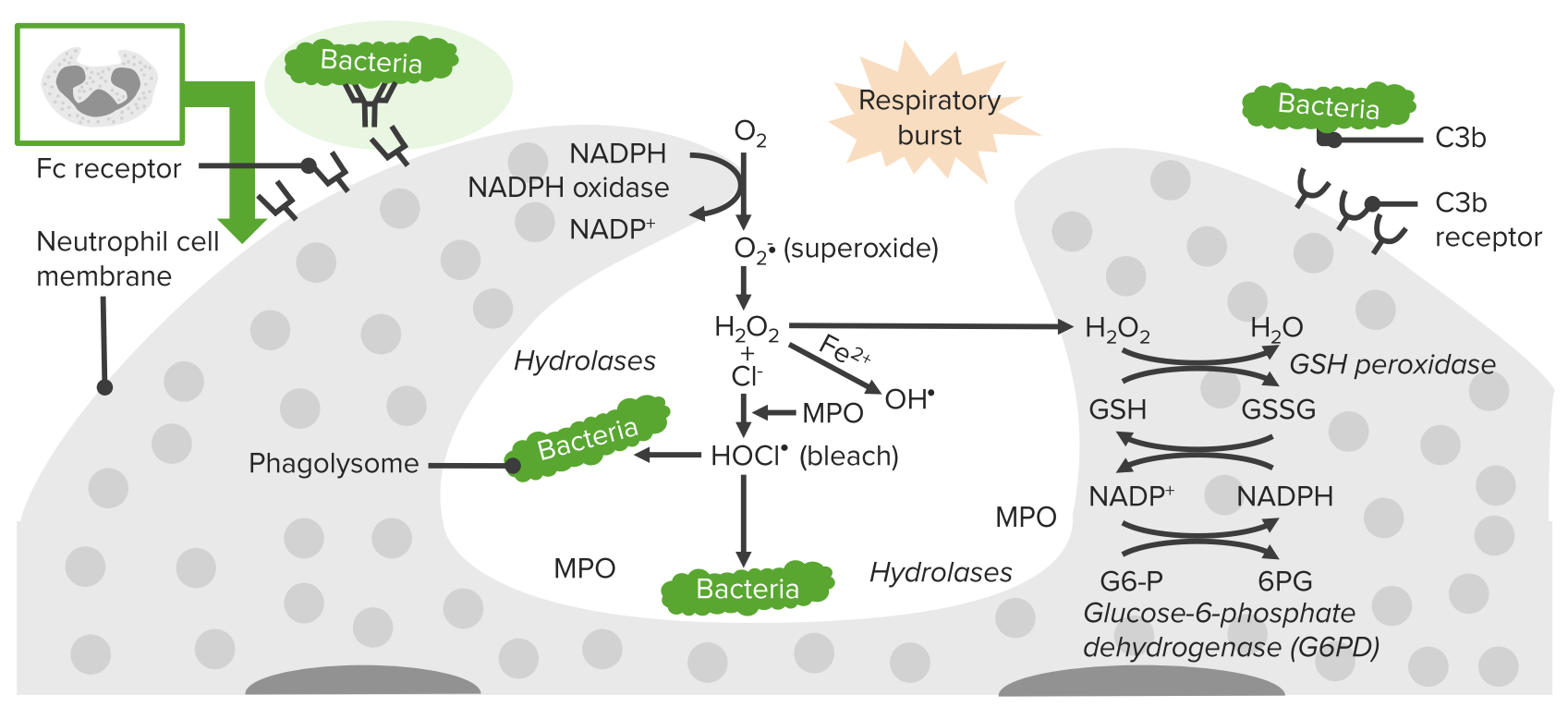
Processo de fagocitose em neutrófilos:
Deficiência na NADPH oxidase resulta na incapacidade de produzir superóxido, o que diminui a capacidade da célula de destruir microrganismos catalase-positivos.
A maioria dos doentes com DGC tem < 5 anos de idade e a gravidade é variável (dependendo do defeito genético). A apresentação clínica pode incluir:
Os doentes podem apresentar infeções repetidas causadas por patogénios bacterianos e fúngicos.
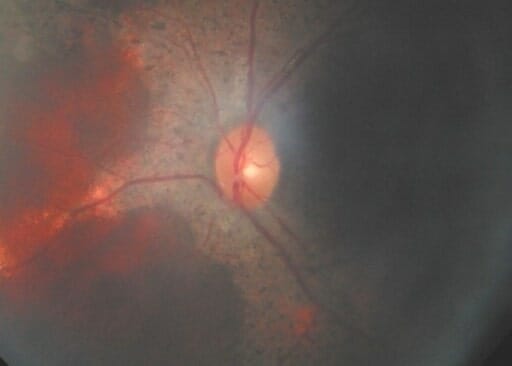
O fundo ótico de um indivíduo com DGC que mostra uma massa na retina
Imagem: “F3: Brother’s fundus” por Mansour A. M. et al. Licença: CC BY 2.0Testes Testes Gonadal Hormones de função de neutrófilos devem ser feitos:
Teste genético molecular e genotipagem:
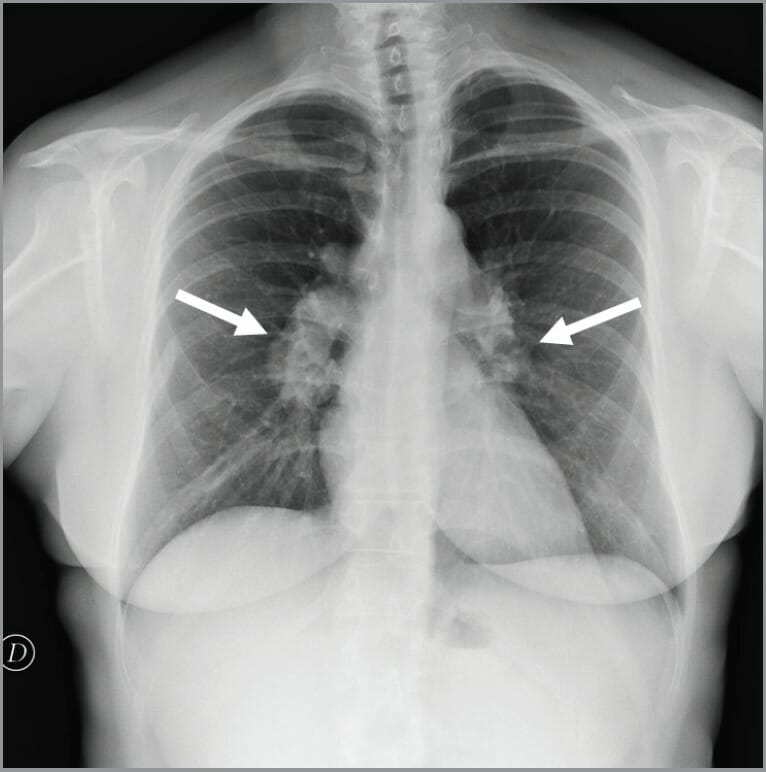
Raio X do toráx que mostra linfadenopatia hilar bilateral num doente com DGC
Imagem: “Chest x-ray showed bilateral hilar lymphadenopathies (arrows) suggestive for chronic granulomatous disease” por Conte G. et al. Licença: CC BY 3.0