Peutz-Jeghers syndrome (PJS) is an autosomal-dominant inherited disorder characterized by GI polyps and mucocutaneous-pigmented macules. Peutz-Jeghers syndrome is 1 of the polyposis syndromes, a group of inherited or acquired conditions characterized by the growth of polyps in the GI tract and associated with other extracolonic features. The syndromes are caused by mutations in specific genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure associated with tumor-suppression or cell-cycle regulation. Peutz-Jeghers syndrome is caused by disruptions in the STK11 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics and is associated with colonic (colorectal) and noncolonic (pancreatic, gastric, breast, uterine, cervical, lung, ovarian, and testicular) cancers. Management is with close surveillance Surveillance Developmental Milestones and Normal Growth and surgery.
Last updated: Jan 18, 2026
Peutz-Jeghers syndrome (PJS) is an autosomal-dominant inherited disorder defined by gastrointestinal, hamartomatous polyps, and skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions/mucosal melanin Melanin Insoluble polymers of tyrosine derivatives found in and causing darkness in skin (skin pigmentation), hair, and feathers providing protection against sunburn induced by sunlight. Carotenes contribute yellow and red coloration. Seborrheic Keratosis macules.
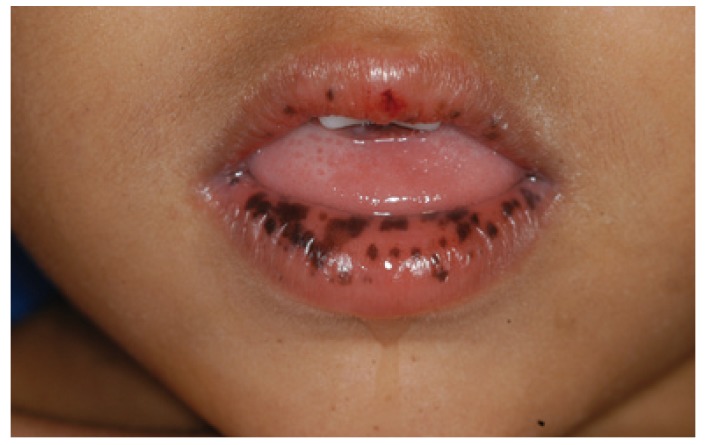
Mucocutaneous pigment macules in a young patient
Image: “Peutz-Jeghers syndrome: black spots localized in the perioral area” by Rogério O. Gondak, et al. License: CC BY 4.0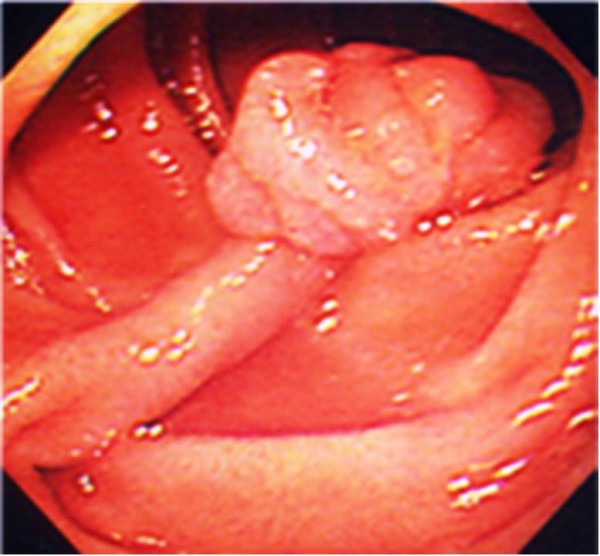
Solitary hamartomatous polyps typical of Peutz-Jeghers syndrome
Image: “Pedunculated duodenal polyp measuring 15 mm in diameter in the second part of the duodenum” by Yusuke Sekino, et al. License: CC BY 2.0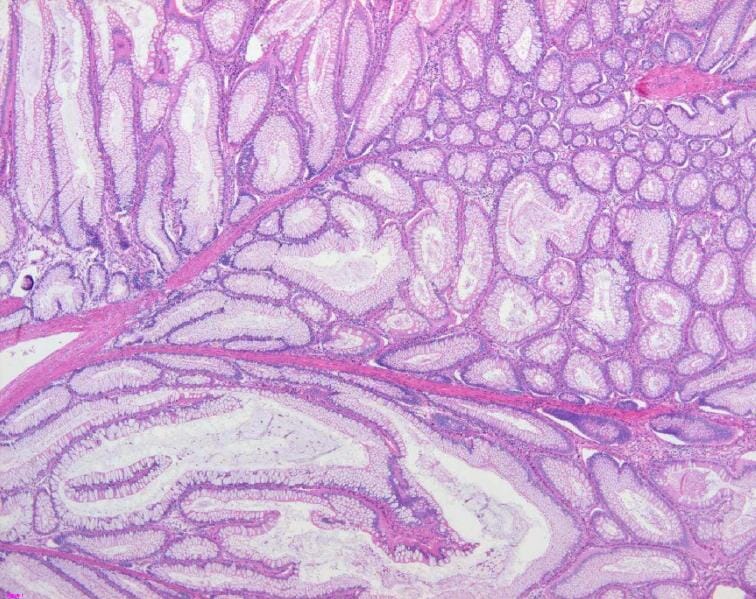
Pathologic findings of a Peutz-Jeghers polyp: The colonic polyp shows hyperplastic mucosal epithelium and an arborizing pattern of smooth muscle, consistent with a hamartomatous polyp (H&E stain, 200x).
Image: “Colon histology with Peutz-Jeghers polyp” by Jong-Ha Yoo et al. License: CC BY 2.0