


A síndrome de Peutz-Jeghers (SPJ) é uma doença hereditária autossómica dominante caracterizada por pólipos gastrointestinais e máculas mucocutâneas pigmentadas. A síndrome de Peutz-Jeghers é uma das síndromes de polipose, um grupo de doenças hereditárias ou adquiridas caracterizadas pelo crescimento de pólipos no trato GI e associadas a outras características extracólicas. São causadas por mutações em genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure específicos associados à supressão tumoral ou à regulação do ciclo celular. A síndrome de Peutz-Jeghers é causada por ruturas no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics STK11 e está associada ao carcinoma do cólon (colorretal) e outros não relacionados com o cólon (pancreático, gástrico, mama, uterino, cervical, pulmonar, ovariano e testicular). O tratamento é feito através de vigilância apertada e cirurgia.
Last updated: Jan 18, 2026
A síndrome de Peutz-Jeghers (SPJ) é uma doença hereditária autossómica dominante caracterizada por pólipos gastrointestinais, hamartomatosos e máculas de melanina na pele/mucosa.
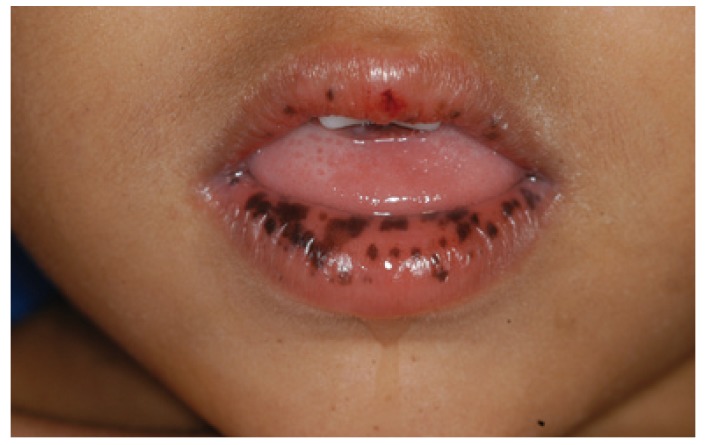
Máculas pigmentares mucocutâneas em doente jovem
Imagem: “Peutz-Jeghers syndrome: black spots localized in the perioral area” por Rogério O. Gondak, et al. Licença: CC BY 4.0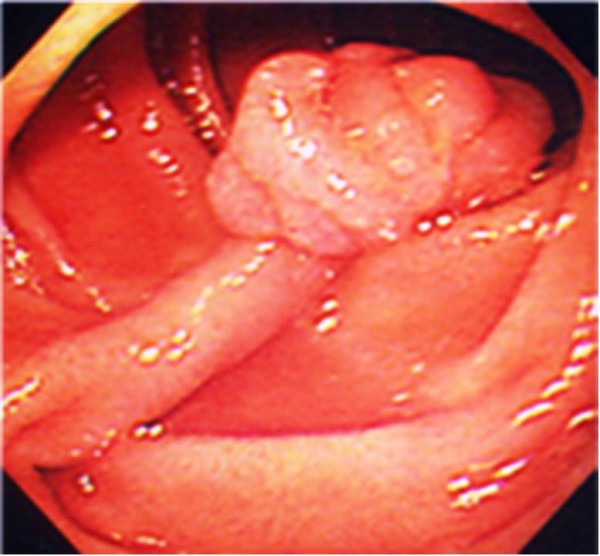
Pólipos hamartomatosos solitários típicos da síndrome de Peutz-Jeghers
Imagem: “Pedunculated duodenal polyp measuring 15 mm in diameter in the second part of the duodenum” por Yusuke Sekino, et al. Licença: CC BY 2.0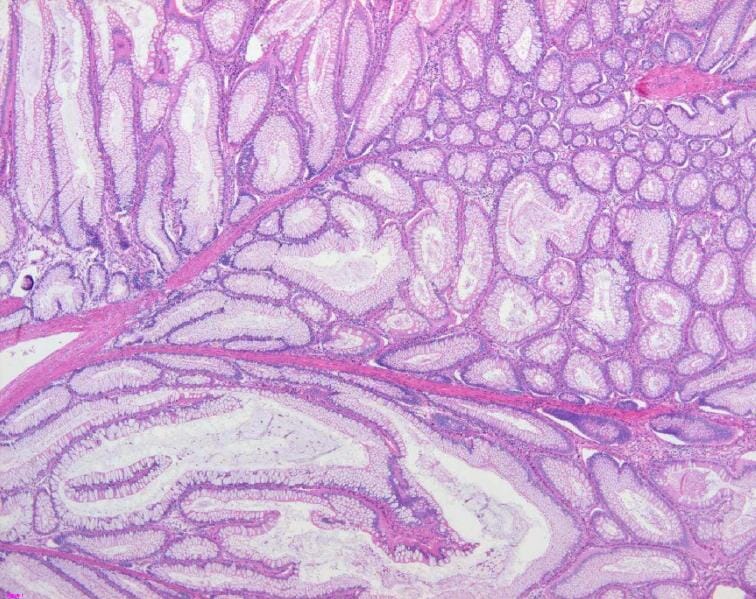
Achados patológicos de um pólipo Peutz-Jeghers: O pólipo intestinal apresenta hiperplasia do epitélio da mucosa e músculo liso com um padrão arborizado, consistente com um pólipo hamartomatoso (coloração H&E, 200x).
Imagem: “Colon histology with Peutz-Jeghers polyp” por Jong-Ha Yoo et al. Licença: CC BY 2.0