Coagulation studies are a group of hematologic laboratory studies that reflect the function of blood vessels, plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology, and coagulation factorsCoagulation factorsEndogenous substances, usually proteins, that are involved in the blood coagulation process.Hemostasis, which all interact with one another to achieve hemostasisHemostasisHemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade.Hemostasis. Coagulation studies are usually ordered to evaluate patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with bleeding or hypercoagulation disorders.
Coagulation studies are a group of hematologic studies that reflect the function of blood vessels, plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology, and coagulation factorsCoagulation factorsEndogenous substances, usually proteins, that are involved in the blood coagulation process.Hemostasis, which all work in harmony to achieve hemostasisHemostasisHemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade.Hemostasis.
Uses of coagulation studies
Evaluation of abnormal bleeding or thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus
The following is a summary of the hemostatic process:
Constriction of the blood vessel: limits blood flowBlood flowBlood flow refers to the movement of a certain volume of blood through the vasculature over a given unit of time (e.g., mL per minute).Vascular Resistance, Flow, and Mean Arterial Pressure to the area
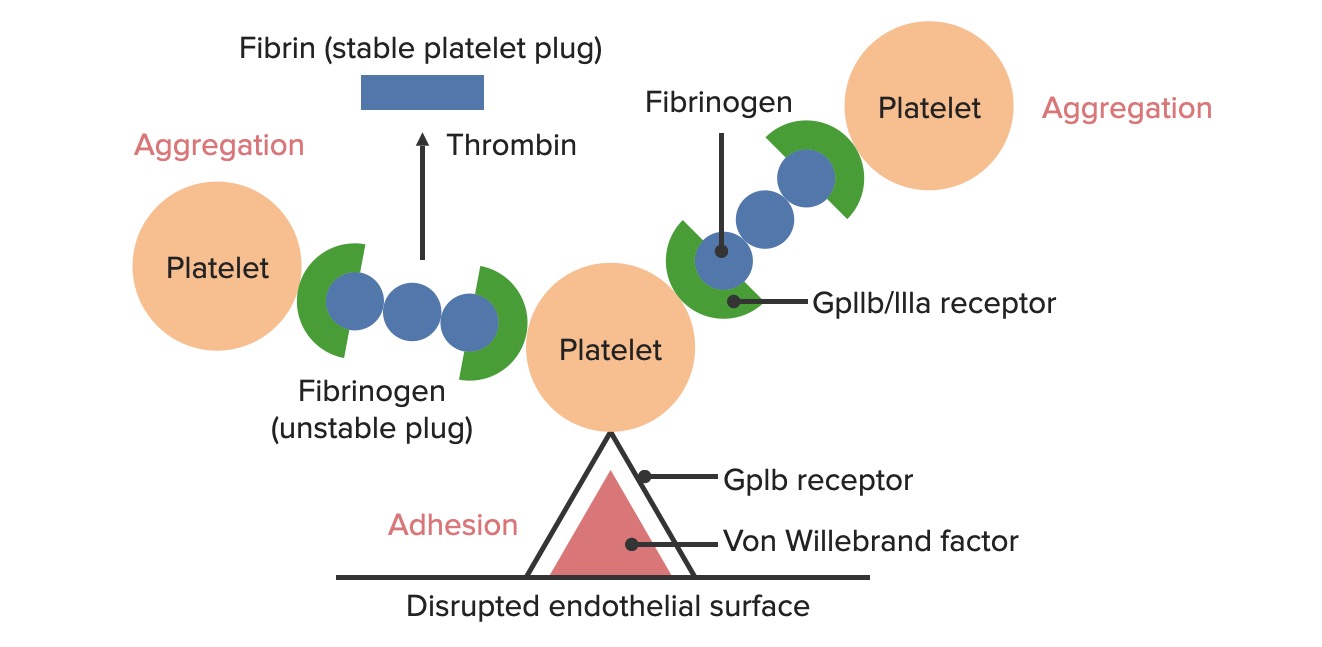
Formation of the platelet plugPlatelet plugHemostasis: the initial, temporary plug formed via the following steps:
Adhesion: exposed von Willebrand factorvon Willebrand factorA high-molecular-weight plasma protein, produced by endothelial cells and megakaryocytes, that is part of the factor VIII/von Willebrand factor complex. The von Willebrand factor has receptors for collagen, platelets, and ristocetin activity as well as the immunologically distinct antigenic determinants. It functions in adhesion of platelets to collagen and hemostatic plug formation. The prolonged bleeding time in von Willebrand diseases is due to the deficiency of this factor.Hemostasis (VWF) binds to the glycoprotein (Gp) Ib receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors on plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Aggregation: GpIIb/IIIa receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors on plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: HistologybindBINDHyperbilirubinemia of the NewbornfibrinogenFibrinogenPlasma glycoprotein clotted by thrombin, composed of a dimer of three non-identical pairs of polypeptide chains (alpha, beta, gamma) held together by disulfide bonds. Fibrinogen clotting is a sol-gel change involving complex molecular arrangements: whereas fibrinogen is cleaved by thrombin to form polypeptides a and b, the proteolytic action of other enzymes yields different fibrinogen degradation products.Hemostasis
Secretion: Substances are released that stimulate further platelet activationPlatelet activationA series of progressive, overlapping events, triggered by exposure of the platelets to subendothelial tissue. These events include shape change, adhesiveness, aggregation, and release reactions. When carried through to completion, these events lead to the formation of a stable hemostatic plug.Hemostasis and aggregation and initiate the coagulation cascadeCoagulation cascadeThe coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot.Hemostasis.
Activation of the coagulation cascadeCoagulation cascadeThe coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot.Hemostasis: forms a more stable fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis clot
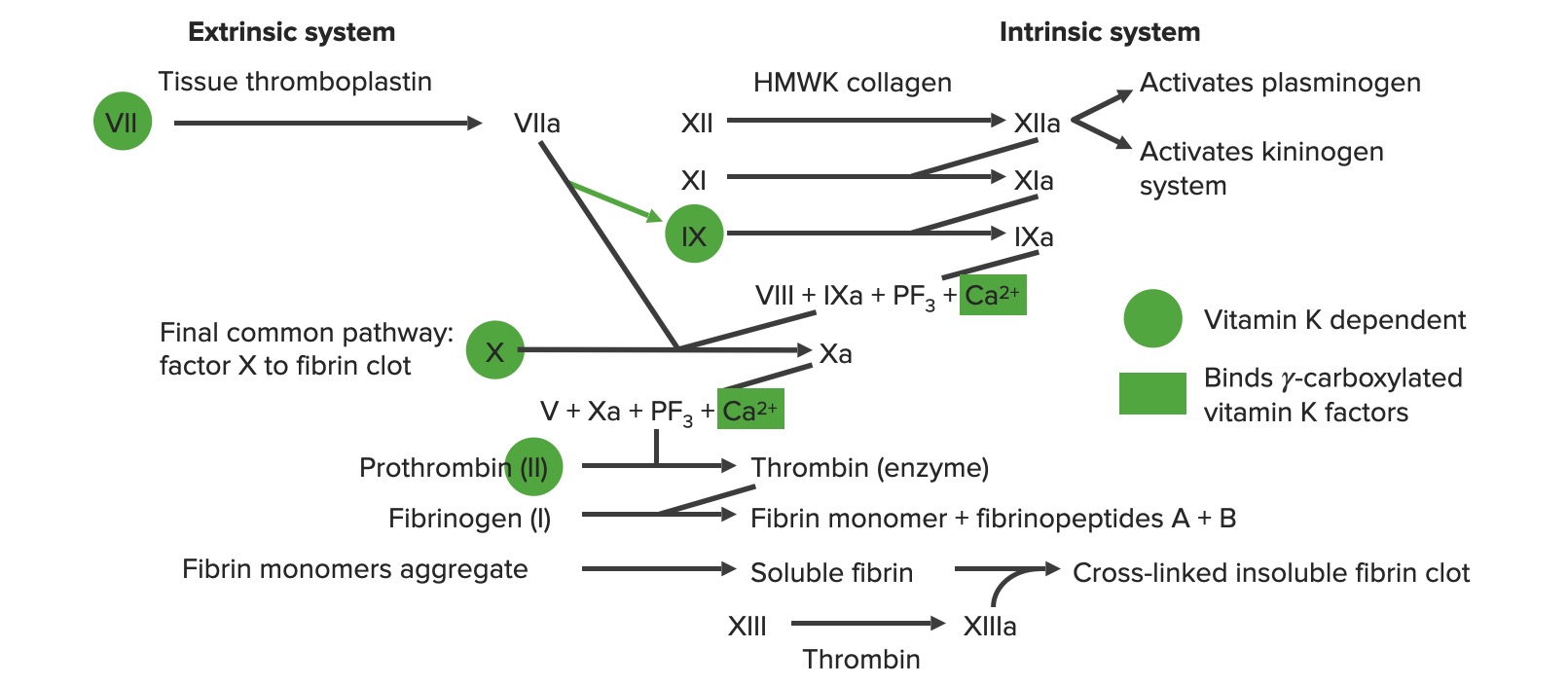
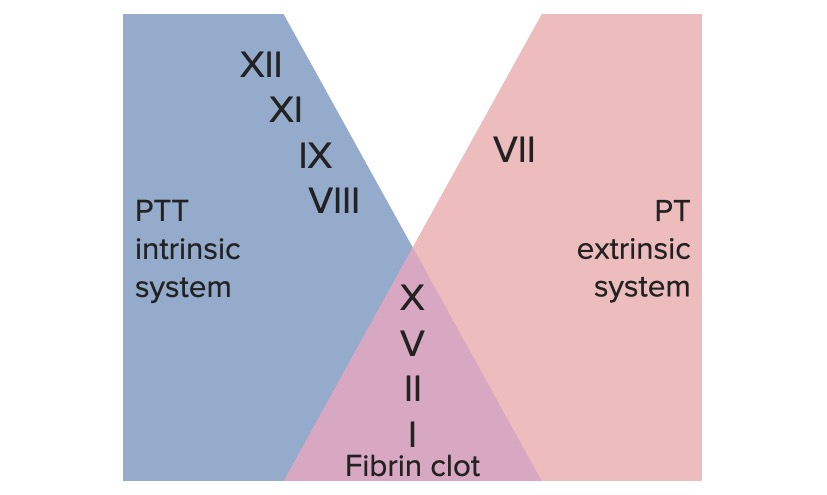
Extrinsic pathwayExtrinsic pathwayThe extrinsic pathway is the primary physiological mechanism by which clotting is initiatedHemostasis:
Primarily responsible for initiation of the cascade
Involves (in order): tissue factor, factor VIIFactor VIIHeat- and storage-stable plasma protein that is activated by tissue thromboplastin to form factor viia in the extrinsic pathway of blood coagulation. The activated form then catalyzes the activation of factor X to factor Xa.Hemostasis, and factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis
Intrinsic pathwayIntrinsic pathwayThe intrinsic pathway is mainly responsible for the amplification of factor X activationHemostasis:
Primarily involved in amplification of the cascade
Can be directly activated by vessel injury
Involves (in order): factors XII, XI, IX, VIII, and X
The extrinsic and intrinsic pathways join together when factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis is activated to form the final common pathwayCommon pathwayHemostasis.
Involves (in order): factors X, V, II (thrombinThrombinAn enzyme formed from prothrombin that converts fibrinogen to fibrin.Hemostasis), I (fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis), and XIII
Inhibition of clotting and fibrinolytic phase:
Stops clotting and breaks down the clot once it is no longer necessary
Involves:
PlasminPlasminA product of the lysis of plasminogen (profibrinolysin) by plasminogen activators. It is composed of two polypeptide chains, light (b) and heavy (a), with a molecular weight of 75, 000. It is the major proteolytic enzyme involved in blood clot retraction or the lysis of fibrin and quickly inactivated by antiplasmins.Hemostasis
AntithrombinAntithrombinEndogenous factors and drugs that directly inhibit the action of thrombin, usually by blocking its enzymatic activity. They are distinguished from indirect thrombin inhibitors, such as heparin, which act by enhancing the inhibitory effects of antithrombins.Anticoagulants
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis C and S
Formation of the temporary hemostatic plug: The disrupted endothelial surface exposes von Willebrand Factor (vWF) to the passing blood. Platelets bind to the vWF via their GpIb receptors and are activated. Platelet activation triggers them to secrete adenosine diphosphate (ADP), which stimulates the expression of the GpIIb/IIIa receptors on the platelets. The GpIIb/IIIa receptors bind to fibrinogen, which is able to bind a platelet on each end, causing platelets to aggregate. As more platelets are bound to one another, the platelet plug is generated. As the coagulation cascade is activated, thrombin converts the weaker fibrinogen into the stronger fibrin, creating a much more stable clot.
Image by Lecturio.
Overview of the coagulation cascade a: activated form PF3: platelet factor 3 (phospholipids)
The following studies can assist in diagnosing issues related to hemostasisHemostasisHemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade.Hemostasis.
Platelet assessments
Platelet counts:
Measure platelet number/concentration
Normal range: 150,000–450,000
↑ in thrombocytosis, which can be caused by:
Acute blood loss/ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements deficiency anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Metastatic cancer
Inflammatory conditions:
Rheumatologic disorders
Inflammatory bowel disease
Kawasaki diseaseKawasaki diseaseAn acute, febrile, mucocutaneous condition accompanied by swelling of cervical lymph nodes in infants and young children. The principal symptoms are fever, congestion of the ocular conjunctivae, reddening of the lips and oral cavity, protuberance of tongue papillae, and edema or erythema of the extremities.Kawasaki Disease
Nephrotic syndromeNephrotic syndromeNephrotic syndrome is characterized by severe proteinuria, hypoalbuminemia, and peripheral edema. In contrast, the nephritic syndromes present with hematuria, variable loss of renal function, and hypertension, although there is sometimes overlap of > 1 glomerular disease in the same individual. Nephrotic Syndrome
InfectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease
Trauma and burnsBurnsA burn is a type of injury to the skin and deeper tissues caused by exposure to heat, electricity, chemicals, friction, or radiation. Burns are classified according to their depth as superficial (1st-degree), partial-thickness (2nd-degree), full-thickness (3rd-degree), and 4th-degree burns. Burns
↓ in thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia:
Immune thrombocytopenic purpuraImmune thrombocytopenic purpuraImmune thrombocytopenic purpura (ITP), formerly known as idiopathic thrombocytopenic purpura, is a condition that develops secondary to immune-mediated destruction of platelets, resulting in thrombocytopenia (platelet count < 100,000/mm³). Immune thrombocytopenic purpura can be either primary or secondary due to drugs or underlying disease. Immune Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of ADAMTS-13, a metalloproteinase that cleaves multimers of von Willebrand factor (VWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets. Thrombotic Thrombocytopenic Purpura
Bleeding timeBleeding timeDuration of blood flow after skin puncture. This test is used as a measure of capillary and platelet function.Hemostasis (BT):
Measures the time for bleeding to stop after a lancet incision
Indirect measure of platelet function
Normal range: 2–7 minutes
Prolonged in:
ThrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia
Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation (DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation)
Von Willebrand diseaseVon Willebrand diseaseVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease (VWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease)
Renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
NSAIDNSAIDNonsteroidal antiinflammatory drugs (NSAIDs) are a class of medications consisting of aspirin, reversible NSAIDs, and selective NSAIDs. NSAIDs are used as antiplatelet, analgesic, antipyretic, and antiinflammatory agents. Nonsteroidal Antiinflammatory Drugs (NSAIDs) and/or aspirinAspirinThe prototypical analgesic used in the treatment of mild to moderate pain. It has anti-inflammatory and antipyretic properties and acts as an inhibitor of cyclooxygenase which results in the inhibition of the biosynthesis of prostaglandins. Aspirin also inhibits platelet aggregation and is used in the prevention of arterial and venous thrombosis.Nonsteroidal Antiinflammatory Drugs (NSAIDs) use
Clotting times
These studies are used to assess the amount of time it takes plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot when various substances are added.
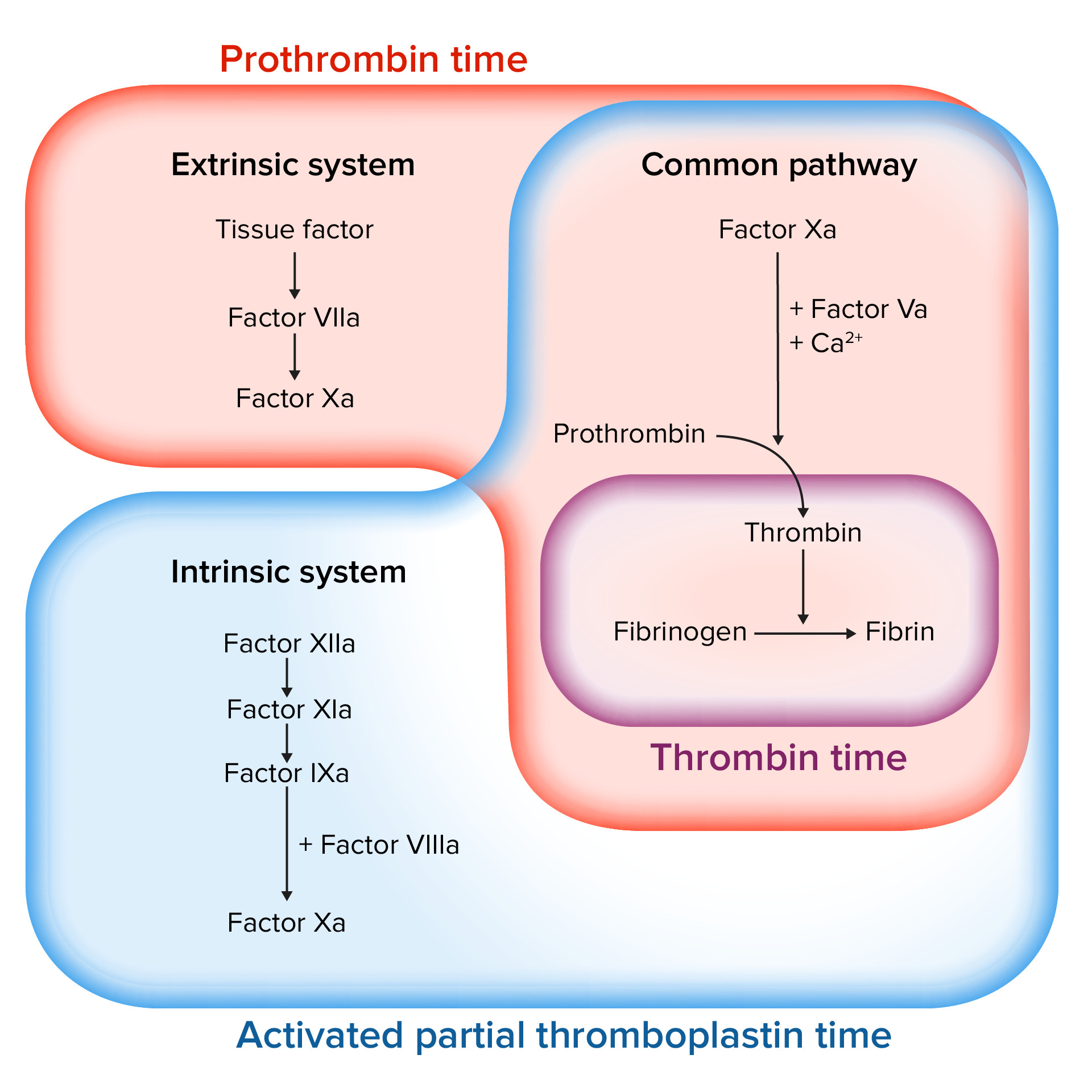
PT:
Measures the time it takes plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot when exposed to tissue factor
Measures function of the extrinsic and commonpathway
Normal range: approximately 11–13 sec
Prolonged in:
WarfarinWarfarinAn anticoagulant that acts by inhibiting the synthesis of vitamin K-dependent coagulation factors. Warfarin is indicated for the prophylaxis and/or treatment of venous thrombosis and its extension, pulmonary embolism, and atrial fibrillation with embolization. It is also used as an adjunct in the prophylaxis of systemic embolism after myocardial infarction. Warfarin is also used as a rodenticide.Anticoagulants therapy
Vitamin K deficiencyVitamin K DeficiencyA nutritional condition produced by a deficiency of vitamin K in the diet, characterized by an increased tendency to hemorrhage (hemorrhagic disorders). Such bleeding episodes may be particularly severe in newborn infants.Fat-soluble Vitamins and their Deficiencies
Factor deficiency: II, V, VII, or X
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
INR:
A ratio comparing the patient’s PT to a reference PT set by the WHO
Measures function of the extrinsic and commonpathways
Normal range: approximately 0.8–1.1
aPTT:
Measures the time it takes plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot when exposed to a negatively charged substance (which activates the intrinsic pathwayIntrinsic pathwayThe intrinsic pathway is mainly responsible for the amplification of factor X activationHemostasis):
Celite
Silica
Measures function of both the intrinsic and commonpathways
Normal range: 25–40 sec
Prolonged in:
Heparin therapy
HemophiliaHemophiliaThe hemophilias are a group of inherited, or sometimes acquired, disorders of secondary hemostasis due to deficiency of specific clotting factors. Hemophilia A is a deficiency of factor VIII, hemophilia B a deficiency of factor IX, and hemophilia C a deficiency of factor XI. Patients present with bleeding events that may be spontaneous or associated with minor or major trauma.Hemophilia disorders (abnormal factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis or IX)
Von Willebrand diseaseVon Willebrand diseaseVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
TT:
Time for plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products to clot after exposure to thrombinThrombinAn enzyme formed from prothrombin that converts fibrinogen to fibrin.Hemostasis
Measures the final step in the common pathwayCommon pathwayHemostasis: the conversion of fibrinogenFibrinogenPlasma glycoprotein clotted by thrombin, composed of a dimer of three non-identical pairs of polypeptide chains (alpha, beta, gamma) held together by disulfide bonds. Fibrinogen clotting is a sol-gel change involving complex molecular arrangements: whereas fibrinogen is cleaved by thrombin to form polypeptides a and b, the proteolytic action of other enzymes yields different fibrinogen degradation products.Hemostasis to fibrinFibrinA protein derived from fibrinogen in the presence of thrombin, which forms part of the blood clot.Rapidly Progressive Glomerulonephritis
Normal range: 14–19 sec
Prolonged in:
↓ FibrinogenFibrinogenPlasma glycoprotein clotted by thrombin, composed of a dimer of three non-identical pairs of polypeptide chains (alpha, beta, gamma) held together by disulfide bonds. Fibrinogen clotting is a sol-gel change involving complex molecular arrangements: whereas fibrinogen is cleaved by thrombin to form polypeptides a and b, the proteolytic action of other enzymes yields different fibrinogen degradation products.Hemostasis
Use of anticoagulantsAnticoagulantsAnticoagulants are drugs that retard or interrupt the coagulation cascade. The primary classes of available anticoagulants include heparins, vitamin K-dependent antagonists (e.g., warfarin), direct thrombin inhibitors, and factor Xa inhibitors. Anticoagulants that inhibit thrombinThrombinAn enzyme formed from prothrombin that converts fibrinogen to fibrin.Hemostasis: heparin, direct thrombinThrombinAn enzyme formed from prothrombin that converts fibrinogen to fibrin.Hemostasis inhibitors (e.g., ArgatrobanArgatrobanAnticoagulants)
DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease
Assessing the coagulation cascade
Image by Lecturio.
Factors involved in the intrinsic, extrinsic, and common pathways
Image by Lecturio.
Coagulation factor assays
Coagulation factor assays primarily used to diagnose specific factor deficiencies. The tests are used to assess the concentration or activity level of individual factors.
Clot-based assays use PT or aPTT testing calibrated for individual factors.
Chromogenic assays use cleavage of a chromogenic (colored) substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes to measure factor activity.
Antigenic assays: ELISAs are used to measure clotting factor levels.
FibrinogenFibrinogenPlasma glycoprotein clotted by thrombin, composed of a dimer of three non-identical pairs of polypeptide chains (alpha, beta, gamma) held together by disulfide bonds. Fibrinogen clotting is a sol-gel change involving complex molecular arrangements: whereas fibrinogen is cleaved by thrombin to form polypeptides a and b, the proteolytic action of other enzymes yields different fibrinogen degradation products.Hemostasis:
Abnormal bleeding typically occurs at levels < 100 mg/dL.
Mixing studies
Mixing studies are used to further evaluate an unexplained prolongation of a clotting test, including a prolonged PT, aPTT, or TT.
Can determine whether an abnormal PT/aPTT is due to:
A factor deficiency OR
Presence of inhibitor:
Autoantibody to a specific factor
Heparins
An elevated C-reactive protein
Measures clotting time of the patient’s plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products when serially diluted with normal plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products
The normal plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products:
Contains all the factors
Does not contain enough factors to overcome the presence of inhibitors
Results: The clotting time will either correct with the addition of normal plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products or fail to correct.
If clotting time (PT or aPTT) corrects:
Diagnosis: factor deficiency
Next step: Identify the specific factor using a coagulation factor assay.
If clotting time (PT or aPTT) fails to correct:
Diagnosis: presence of a factor inhibitor
Next steps:
Consult with hematology.
Review medications for potential cause.
Determine the titer of antibody present using further serial dilutions.
HypocoagulableHypocoagulableHypocoagulable conditions, also known as bleeding disorders or bleeding diathesis, are a diverse group of diseases that result in abnormal hemostasis. Physiologic hemostasis is dependent on the integrity of endothelial cells, subendothelial matrix, platelets, and coagulation factors. The hypocoagulable states result from abnormalities in one or more of these contributors, resulting in ineffective thrombosis and bleeding.Hypocoagulable Conditions conditions are a group of diseases that result in abnormal hemostasisHemostasisHemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade.Hemostasis. They manifest with minor bleeding (petechiaePetechiaePrimary Skin Lesions, mucocutaneous bleeding) or more severe internal bleeding (hemarthrosisHemarthrosisBleeding into the joints. It may arise from trauma or spontaneously in patients with hemophilia.Hemophilia, intracranial bleeding).
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with these disorders will most likely have an increased bleeding timeBleeding timeDuration of blood flow after skin puncture. This test is used as a measure of capillary and platelet function.Hemostasis but normal PT and aPTT.
Glanzmann thrombastheniaGlanzmann ThrombastheniaHypocoagulable Conditions: an autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance bleeding syndrome characterized by a deficiency of the GpIIb/IIIa receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, resulting in a lack of platelet aggregationPlatelet aggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Hemostasis
Bernard–Soulier syndrome: an autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance bleeding syndrome characterized by deficiency of the GpIb receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, resulting in failure of platelet adhesionPlatelet adhesionExposure of the blood to subendothelial components at the site of injury causes platelets to adhere to the injury site.Hemostasis: Bernard–Soulier syndrome can be diagnosed with a ristocetin assay. Ristocetin activates VWF to allow binding to the platelet GpIb receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors; however, in Bernard–Soulier syndrome, plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology will fail to adhere with the assay.
ThrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia: When platelet levels are low, formation of the platelet plugPlatelet plugHemostasis may be impaired. ThrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia is usually diagnosed with a CBC with platelet count and morphology assessment. Causes may include immune thrombocytopenic purpuraImmune thrombocytopenic purpuraImmune thrombocytopenic purpura (ITP), formerly known as idiopathic thrombocytopenic purpura, is a condition that develops secondary to immune-mediated destruction of platelets, resulting in thrombocytopenia (platelet count < 100,000/mm³). Immune thrombocytopenic purpura can be either primary or secondary due to drugs or underlying disease. Immune Thrombocytopenic Purpura (ITPITPImmune thrombocytopenic purpura (ITP), formerly known as idiopathic thrombocytopenic purpura, is a condition that develops secondary to immune-mediated destruction of platelets, resulting in thrombocytopenia (platelet count < 100,000/mm³). Immune thrombocytopenic purpura can be either primary or secondary due to drugs or underlying disease. Immune Thrombocytopenic Purpura), thrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpuraThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of ADAMTS-13, a metalloproteinase that cleaves multimers of von Willebrand factor (VWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets. Thrombotic Thrombocytopenic Purpura (TTPTTPThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of adamts-13, a metalloproteinase that cleaves multimers of von Willebrand factor (vWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets.Thrombotic Thrombocytopenic Purpura), HIVHIVAnti-HIV Drugs, drug-induced thrombocytopeniaDrug-Induced ThrombocytopeniaThrombocytopenia, or other bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis disorders (including malignancyMalignancyHemothorax).
Disorders of the coagulation cascadeCoagulation cascadeThe coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot.Hemostasis
HemophiliaHemophiliaThe hemophilias are a group of inherited, or sometimes acquired, disorders of secondary hemostasis due to deficiency of specific clotting factors. Hemophilia A is a deficiency of factor VIII, hemophilia B a deficiency of factor IX, and hemophilia C a deficiency of factor XI. Patients present with bleeding events that may be spontaneous or associated with minor or major trauma.Hemophilia: a rare blood clotting disorder in which the body lacks blood-clotting factors (factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis in hemophilia AHemophilia AThe classic hemophilia resulting from a deficiency of factor VIII. It is an inherited disorder of blood coagulation characterized by a permanent tendency to hemorrhage.Hemophilia and factor IXFactor IXStorage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa.Hemostasis in hemophilia BHemophilia BA deficiency of blood coagulation factor IX inherited as an X-linked disorder. (also known as Christmas disease, after the first patient studied in detail, not the holiday.) historical and clinical features resemble those in classic hemophilia, but patients present with fewer symptoms. Severity of bleeding is usually similar in members of a single family. Many patients are asymptomatic until the hemostatic system is stressed by surgery or trauma. Treatment is similar to that for hemophilia A.Hemophilia). Affected individuals present with abnormal bleeding that occurred spontaneously or after minor trauma. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship can bleed into joint spaces and can develop life-threatening internal bleeding. Coagulation studies typically reveal a prolonged aPTT due to the abnormalities in the intrinsic pathwayIntrinsic pathwayThe intrinsic pathway is mainly responsible for the amplification of factor X activationHemostasis, but the PT and platelet counts are normal. Mixing studies will correct, and the level of the specific factor activity in which the patient is deficient will be low.
Mixed disorders affecting both plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology and coagulation factorsCoagulation factorsEndogenous substances, usually proteins, that are involved in the blood coagulation process.Hemostasis
Von Willebrand diseaseVon Willebrand diseaseVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease (VWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease): the most commonly inherited disorder of hemostasisHemostasisHemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade.Hemostasis, caused by a qualitative or quantitative deficiency in VWF: The three primary types of VWDvWDVon Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease differ in severity, though all tend to present with bleeding abnormalities. Von Willebrand factorvon Willebrand factorA high-molecular-weight plasma protein, produced by endothelial cells and megakaryocytes, that is part of the factor VIII/von Willebrand factor complex. The von Willebrand factor has receptors for collagen, platelets, and ristocetin activity as well as the immunologically distinct antigenic determinants. It functions in adhesion of platelets to collagen and hemostatic plug formation. The prolonged bleeding time in von Willebrand diseases is due to the deficiency of this factor.Hemostasis is required for both initial platelet adhesionPlatelet adhesionExposure of the blood to subendothelial components at the site of injury causes platelets to adhere to the injury site.Hemostasis, and it helps stabilize factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis in the intrinsic pathwayIntrinsic pathwayThe intrinsic pathway is mainly responsible for the amplification of factor X activationHemostasis. Testing shows a normal platelet count, but the aPTT may be prolonged (depending on the reduction of factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis activity). A VWF antigenAntigenSubstances that are recognized by the immune system and induce an immune reaction.Vaccination test, specific functional assays, and genetic testingGenetic TestingDetection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing.Myotonic Dystrophies can aid in the diagnosis.
Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation: a serious medical condition in which the coagulation cascadeCoagulation cascadeThe coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot.Hemostasis is activated systemically, leading to multiple clots that can lead to permanent end-organ damage and in the process, coagulation factorsCoagulation factorsEndogenous substances, usually proteins, that are involved in the blood coagulation process.Hemostasis are used up: Disseminated intravascular coagulationDisseminated intravascular coagulationDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation always has a secondary cause. InfectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, burnsBurnsA burn is a type of injury to the skin and deeper tissues caused by exposure to heat, electricity, chemicals, friction, or radiation. Burns are classified according to their depth as superficial (1st-degree), partial-thickness (2nd-degree), full-thickness (3rd-degree), and 4th-degree burns. Burns, and malignancies are among the most common causes, but DICDICDisseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation can also occur during severe postpartum hemorrhagePostpartum hemorrhagePostpartum hemorrhage is one of the most common and deadly obstetric complications. Since 2017, postpartum hemorrhage has been defined as blood loss greater than 1,000 mL for both cesarean and vaginal deliveries, or excessive blood loss with signs of hemodynamic instability. Postpartum Hemorrhage. Laboratory findings include thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, prolongation of the PT and aPTT, and elevation of D-dimerD-dimerDeep Vein Thrombosis.
CirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis: The liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy is the primary site of synthesisSynthesisPolymerase Chain Reaction (PCR) for a majority of the clotting factors. In addition to impaired synthesisSynthesisPolymerase Chain Reaction (PCR) of clotting factors, cirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis also may independently result in thrombocytopeniaThrombocytopeniaThrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia due to splenic sequestrationSplenic sequestrationSevere Congenital Neutropenia of plateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology and decreased thrombopoietinThrombopoietinA humoral factor that stimulates the production of thrombocytes (blood platelets). Thrombopoietin stimulates the proliferation of bone marrow megakaryocytes and their release of blood platelets. The process is called thrombopoiesis.Platelets: Histology production. PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology themselves may also be dysfunctional. The platelet count may be low and the PT and aPTT may both be prolonged.
References
Longo, D., Fauci, A., Kasper, D., Hauser, S., Jameson, J., Loscalzo, J. Harrison’s Manual of Medicine, 18th ed. McGraw-Hill Professional, 2012, pp 2159–2238.