Immune thrombocytopenic purpura (ITP), formerly known as idiopathic Idiopathic Dermatomyositis thrombocytopenic purpura, is a condition that develops secondary to immune-mediated destruction of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology, resulting in thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia (platelet count < 100,000/mm³). Immune thrombocytopenic purpura can be either primary or secondary due to drugs or underlying disease. The diagnosis is usually one of exclusion. Many patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with ITP are asymptomatic. When present, symptoms are primarily related to bleeding (e.g., bruising, petechiae Petechiae Primary Skin Lesions, epistaxis Epistaxis Bleeding from the nose. Granulomatosis with Polyangiitis), but fatigue Fatigue The state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli. Fibromyalgia is also common. The severity of thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia in patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with ITP is variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables. When platelet counts Platelet counts The number of platelets per unit volume in a sample of venous blood. Coagulation Studies drop to < 20,000/mm³, the risk of serious bleeding increases. Treatment may include platelet transfusion, steroids Steroids A group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus. Benign Liver Tumors, IV immune globulins, and/or splenectomy Splenectomy Surgical procedure involving either partial or entire removal of the spleen. Rupture of the Spleen. Some cases remit spontaneously; others generally have a good prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas with appropriate therapy.
Last updated: Dec 15, 2025
Immune thrombocytopenic purpura (ITP) is an acquired thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia that results from autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques targeting platelet antigens.
Onset:
Clinical manifestations:
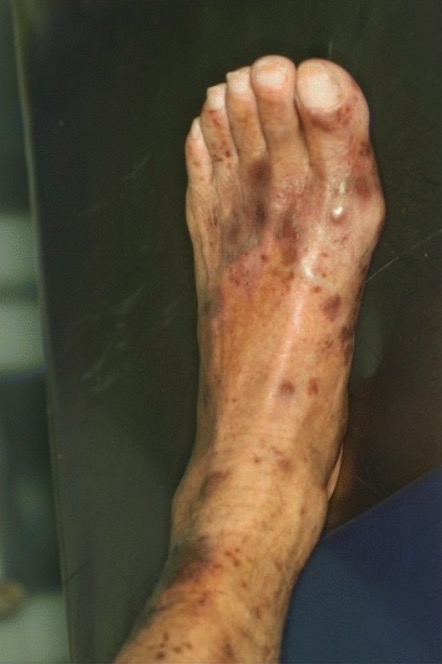
Immune thrombocytopenic purpura (ITP):
Skin purpuric lesions
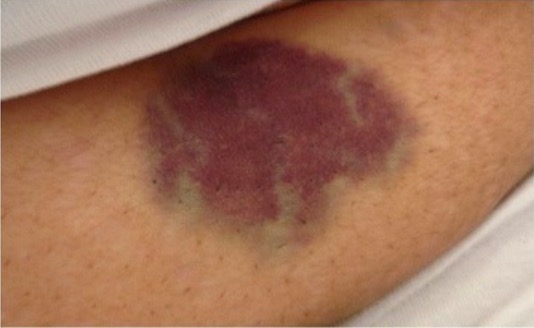
Abdominal ecchymosis in a bee-venom induced immune thrombocytopenia
Image: “Abdominal ecchymosis size” by Department of Internal Medicine, Mubarak Al-Kabeer Hospital, P.O. Box 800, Dasman, 15458, Kuwait. License: CC BY 4.0Indications:
Mild bleeding:
Severe bleeding (GI/intracranial; platelet count: < 10,000/mm³):
Additional therapies for active/persistent bleeding:
No bleeding and platelet count > 30,000/mm³:
Avoid:
Recurrent/refractory episodes:
Post-splenectomy ITP/refractory to steroids Steroids A group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus. Benign Liver Tumors: