La púrpura trombocitopénica inmune (PTI), antes conocida como púrpura trombocitopénica idiopática, es una afección que se desarrolla de forma secundaria a la destrucción inmune de las plaquetas, lo que provoca trombocitopenia (recuento de plaquetas < 100 000/mm³). La púrpura trombocitopénica inmune puede ser primaria o secundaria debido a medicamentos o enfermedades subyacentes. El diagnóstico suele ser de exclusión. Muchos pacientes con PTI son asintomáticos. Cuando se presentan, los LOS Neisseria síntomas están relacionados principalmente con hemorragias (e.g., hematomas, petequias, epistaxis Epistaxis Bleeding from the nose. Granulomatosis with Polyangiitis), pero también es frecuente la fatiga. La gravedad de la trombocitopenia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria pacientes con PTI es variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables. Cuando el recuento de plaquetas desciende a < 20 000/mm³, aumenta el riesgo de hemorragias graves. El tratamiento puede incluir transfusiones de plaquetas, esteroides, inmunoglobulinas intravenosas y/o esplenectomía. Algunos casos remiten espontáneamente; otros tienen generalmente un buen pronóstico con una terapia adecuada.
Last updated: Dec 15, 2025
La púrpura trombocitopénica inmune es una trombocitopenia adquirida que resulta de autoanticuerpos dirigidos a antígenos plaquetarios.
Inicio:
Manifestaciones clínicas:
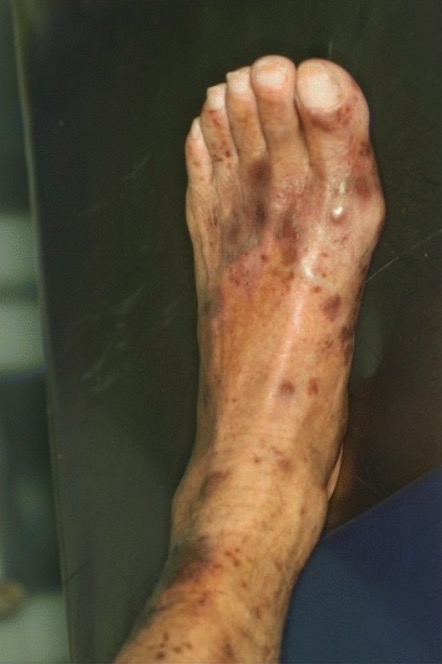
Púrpura trombocitopénica inmune (PTI):
Lesiones cutáneas purpúricas
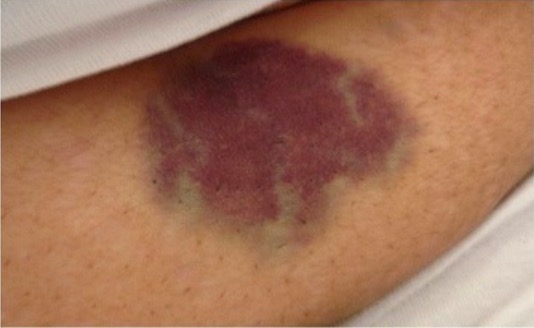
Equimosis abdominal en una trombocitopenia inmune inducida por el veneno de abeja
Imagen: “Abdominal ecchymosis size” por Department of Internal Medicine, Mubarak Al-Kabeer Hospital, P.O. Box 800, Dasman, 15458, Kuwait. Licencia: CC BY 4.0Indicaciones:
Hemorragia leve:
Hemorragia grave (gastrointestinal/intracraneal; recuento de plaquetas: < 10 000/mm³):
Terapias adicionales para hemorragias activas/persistentes:
Sin hemorragia y recuento de plaquetas > 30 000/mm³:
Evitar:
Episodios recurrentes/refractarios:
PTI post-esplenectomía/refractaria a esteroides: