Hypersensitivity pneumonitis Pneumonitis Human Herpesvirus 6 and 7 (HP), previously called extrinsic allergic alveolitis, is an immunologically induced inflammatory disease affecting the alveoli Alveoli Small polyhedral outpouchings along the walls of the alveolar sacs, alveolar ducts and terminal bronchioles through the walls of which gas exchange between alveolar air and pulmonary capillary blood takes place. Acute Respiratory Distress Syndrome (ARDS), bronchioles Bronchioles The small airways branching off the tertiary bronchi. Terminal bronchioles lead into several orders of respiratory bronchioles which in turn lead into alveolar ducts and then into pulmonary alveoli. Bronchial Tree: Anatomy, and lung parenchyma. It is caused by repeated inhalation of an inciting agent in a susceptible host that triggers first a type III (immune complex-mediated) hypersensitivity reaction in the acute phase Acute phase Short Bowel Syndrome and then a type IV (delayed) reaction in the subacute and chronic phases. The clinical presentation of acute HP includes cough, fever Fever Fever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever, and malaise Malaise Tick-borne Encephalitis Virus, while subacute and chronic forms present as insidious onset of a cough and dyspnea Dyspnea Dyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea over weeks to months. Diagnosis is aided by high-resolution CT High-resolution CT Imaging of the Lungs and Pleura scans and bronchoalveolar lavage Bronchoalveolar lavage Washing out of the lungs with saline or mucolytic agents for diagnostic or therapeutic purposes. It is very useful in the diagnosis of diffuse pulmonary infiltrates in immunosuppressed patients. Pulmonary Fibrosis. Management includes avoiding the inciting agent and administration of steroids Steroids A group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus. Benign Liver Tumors in subacute and chronic cases. Early treatment has a good prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas, but long-term exposure can cause permanent scarring Scarring Inflammation and fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans.
Last updated: Dec 15, 2025
Hypersensitivity pneumonitis Pneumonitis Human Herpesvirus 6 and 7 (HP), also called extrinsic allergic alveolitis, is an immunologically induced inflammatory disease affecting the lung parenchyma, alveoli Alveoli Small polyhedral outpouchings along the walls of the alveolar sacs, alveolar ducts and terminal bronchioles through the walls of which gas exchange between alveolar air and pulmonary capillary blood takes place. Acute Respiratory Distress Syndrome (ARDS), and bronchioles Bronchioles The small airways branching off the tertiary bronchi. Terminal bronchioles lead into several orders of respiratory bronchioles which in turn lead into alveolar ducts and then into pulmonary alveoli. Bronchial Tree: Anatomy. This disorder is caused by repeated inhalation of inciting agents in a susceptible host.
Interstitial lung disease caused by hypersensitivity reactions:
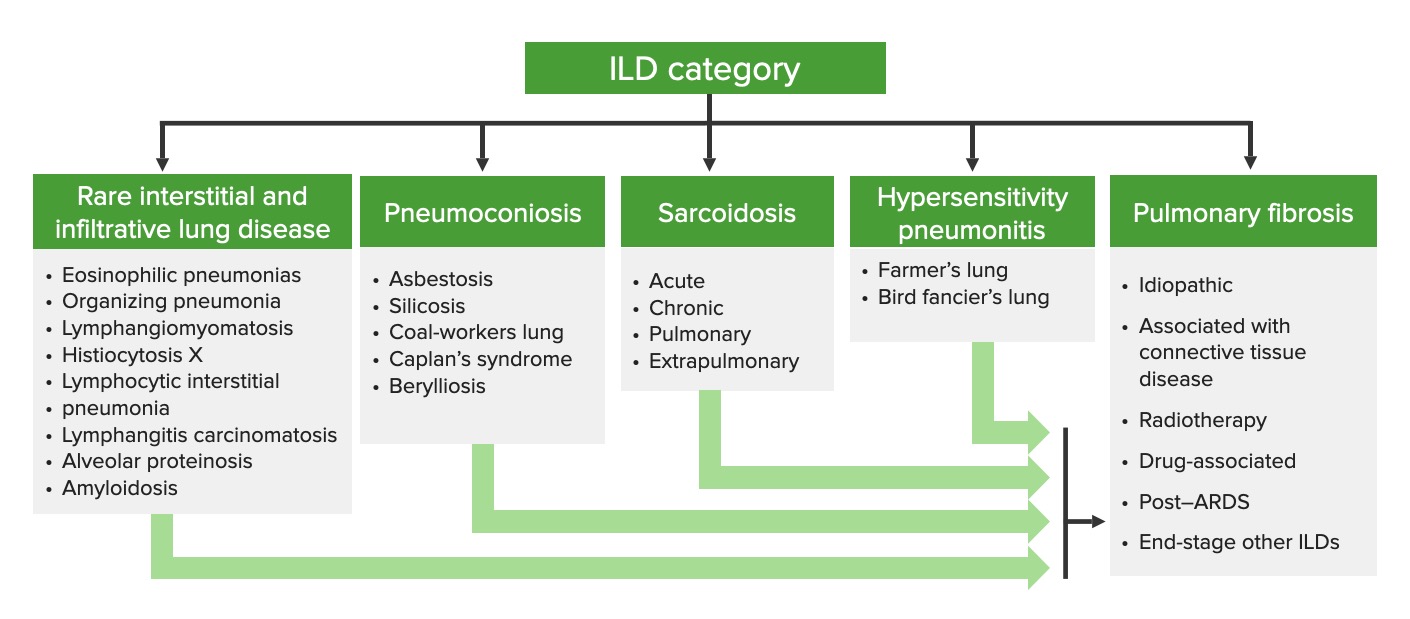
Categorization of interstitial lung diseases
ILD: interstitial lung disease
Classification is based on duration of illness and frequency, duration, and intensity of exposure. There is considerable variability in presentation and course.
More than 300 etiologies of HP have been reported across a wide range of exposures involving airborne antigens.
| Disease | Exposure | Antigens |
|---|---|---|
| Farmer’s lung |
|
|
| Bird fancier’s lung |
|
Proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis present on feathers and in excreta |
| Chemical worker’s lung | Many different chemicals | Isocyanates |
| Byssinosis | Working in the textile industry |
|
| Bagassosis | Moldy sugar cane | Thermophilic Thermophilic Campylobacter actinomycetes |
| Silo filler’s lung | Fermentation in silos | Nitrogen Nitrogen An element with the atomic symbol n, atomic number 7, and atomic weight [14. 00643; 14. 00728]. Nitrogen exists as a diatomic gas and makes up about 78% of the earth’s atmosphere by volume. It is a constituent of proteins and nucleic acids and found in all living cells. Urea Cycle dioxide gas |
| Maltworker’s lung | Turning germinating barley |
|
| Humidifier fever Fever Fever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever | Contaminated humidifying systems in air conditioners |
|
| Mushroom workers lung | Turning mushroom compost | Thermophilic Thermophilic Campylobacter actinomycetes |
| Cheese washer’s lung | Moldy cheese | Penicillin Penicillin Rheumatic Fever casei |
| Winemaker’s lung | Mold Mold Mycology on grapes | Botrytis |
Initially during acute HP, the disease process is driven by a type III (immune-complex–mediated) hypersensitivity reaction. Continued exposure shifts the process to a type IV delayed hypersensitivity reaction Delayed Hypersensitivity Reaction Glycopeptides mediated by T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified – cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions.
Pathogenesis is directly related to antibodies Antibodies Immunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution. Immunoglobulins: Types and Functions targeting specific protein antigens:
Acute HP (type III immune-complex–mediated hypersensitivity reaction):
Subacute and chronic HP (type IV delayed hypersensitivity reaction Delayed Hypersensitivity Reaction Glycopeptides mediated by T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified – cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions):
Acute HP usually follows massive exposure to an inciting agent:

Hypersensitivity pneumonitis (HP):
The cellular interstitium of subacute HP (A) is dominated by plasma cells (magnification in B). A typical poorly formed granuloma of HP is present in (B).
(A,B) H&E stain; (A) 40× original magnification; (B) 400× original magnification
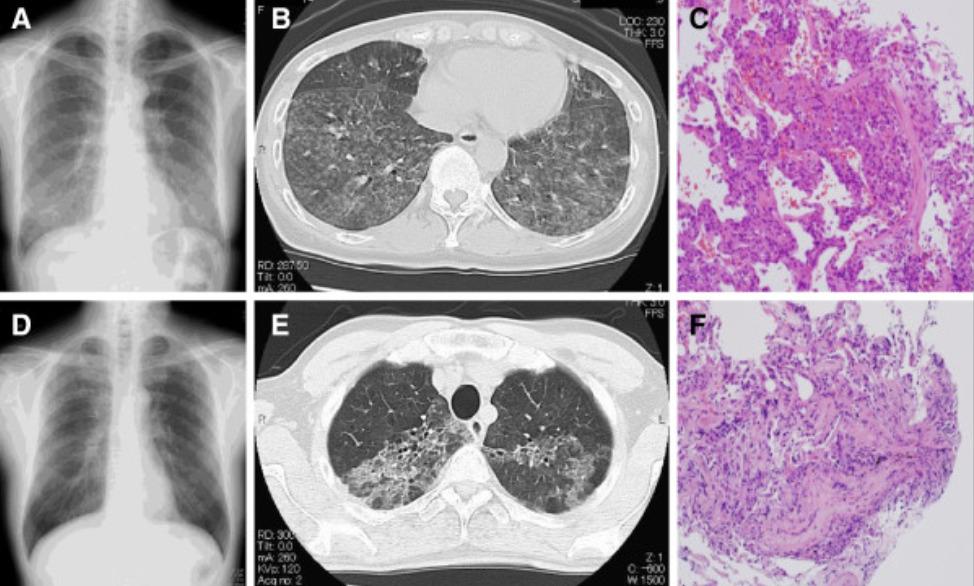
Radiological and pathological assessment for a husband and wife in Japan who both developed hypersensitivity pneumonitis caused by inhalation of Trichosporon asahii which was growing in the straw mats of their home.
Wife: Chest x-ray on the day of admission shows mild infiltrates predominantly in the middle to lower lung fields with multiple scattered nodular lesions (Panel A).
Thoracic CT (computed tomography) taken on the same day confirms that these lesions correspond to ground-glass opacities and abundant centrilobular nodules, predominantly located in the bilateral lower lobes (Panel B).
Specimens obtained from transbronchial lung biopsy (TBLB) demonstrate organization within the peribronchial area with alveolitis, suggesting transbronchial spread (Panel C).
Husband: Chest x-ray (Panel D) and thoracic CT (Panel E) show ground glass opacities in the upper lungs on both sides.
The specimens obtained from TBLB at his local hospital reveals organizing tissue within the peribronchial area with alveolitis, suggesting transbronchial spread (Panel F).
Exact diagnostic criteria are still being debated but generally include the following: