Autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance hyper-IgE syndrome (AD-HIES), also known as Job's syndrome, is a rare form of primary immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome disorder that affects various organs systems in addition to the immune system Immune system The body's defense mechanism against foreign organisms or substances and deviant native cells. It includes the humoral immune response and the cell-mediated response and consists of a complex of interrelated cellular, molecular, and genetic components. Primary Lymphatic Organs. Some cases of AD-HIES are caused by mutations in the STAT3 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics, resulting in abnormal neutrophil chemotaxis Chemotaxis The movement of leukocytes in response to a chemical concentration gradient or to products formed in an immunologic reaction. Leukocyte Adhesion Deficiency Type 1. In other cases, the cause is unknown. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with AD-HIES experience recurrent pneumonia Pneumonia Pneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy. Pneumonia, skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, rashes Rashes Rashes are a group of diseases that cause abnormal coloration and texture to the skin. The etiologies are numerous but can include irritation, allergens, infections, or inflammatory conditions. Rashes that present in only 1 area of the body are called localized rashes. Generalized rashes occur diffusely throughout the body. Generalized and Localized Rashes, blisters, and abscesses.
Last updated: Dec 15, 2025
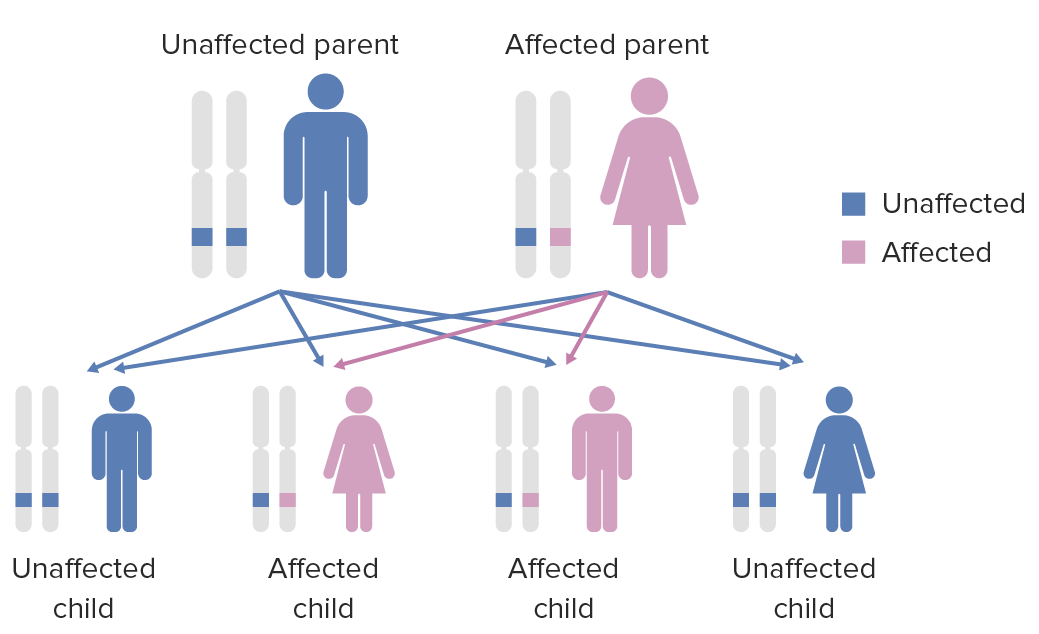
Diagram of the inheritance pattern of autosomal dominant conditions
Image by Lecturio.Symptoms may be apparent at birth or become apparent during infancy or early childhood.
Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with various clinical manifestations and abnormalities:

Patient with characteristic facial features of Job’s syndrome. Note the broad nose, deep-set-eyes, prognathism, and rough facial skin with exaggerated pore size.
Image: “Facial features in AD-HIES” by Milton S. Hershey Medical Center, School of Medicine, Hershey, PA. License: CC BY 2.0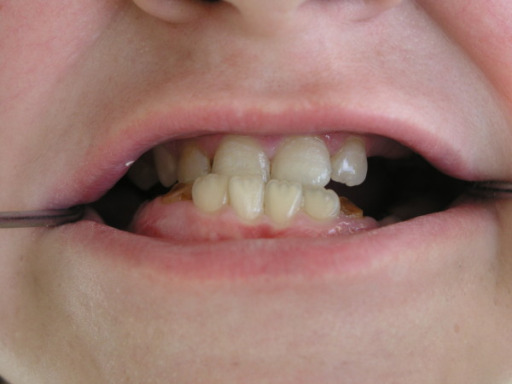
Prognathism in a child with AD-HIES
Image: “Prognathism in a child with HIES” by Department of Pediatric Pneumonology, Allergology and Clinical Immunology, Poznan University of Medical Sciences, 27/33 Szpitalna Street, Poznan, Poland. License: CC BY 2.0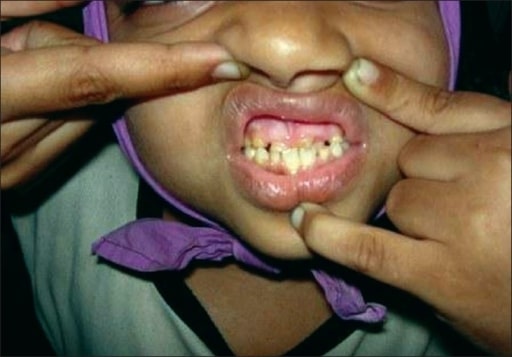
Photograph of a 10-year-old girl with AD-HIES, revealing retention of primary dentition
Image: “Retention of primary dentition” by Department of Dermatology, Madras Medical College, Chennai, India. License: CC BY 2.0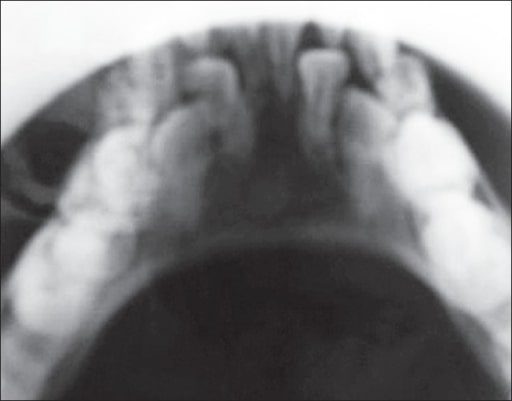
X-ray of a ten-year-old girl with AD-HIES, revealing retention of primary dentition
Image: “Jaw X-ray showing retention of primary dentition” by Department of Dermatology, Madras Medical College, Chennai, India. License: CC BY 2.0A helpful mnemonic of the clinical manifestations of AD-HIES is FATED:
The diagnosis of AD-HIES is based on clinical evaluation, especially a detailed patient history and identification Identification Defense Mechanisms of characteristic findings.
There is no definitive cure. Management is aimed at treating symptoms as well as preventing and managing infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease.
The following conditions are differential diagnoses of autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance hyperimmunoglobulin E syndrome: