Acromegaly and gigantism are caused by an excessive production of growth hormone (GH) by the pituitary gland. Gigantism typically results from excess GH before closure of the growth plate in children. Acromegaly typically results from excess GH after closure of the growth plate. Tall stature is a clinical sign observed in some patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with gigantism. Increased GH and insulin-like growth factor-1 Insulin-like growth factor-1 A well-characterized basic peptide believed to be secreted by the liver and to circulate in the blood. It has growth-regulating, insulin-like, and mitogenic activities. This growth factor has a major, but not absolute, dependence on growth hormone. It is believed to be mainly active in adults in contrast to insulin-like growth factor II, which is a major fetal growth factor. Hypertrophic and Keloid Scars (IGF-1) are responsible for inducing hypersomatotropism. Acromegaly is usually produced by pituitary tumors Pituitary tumors Neoplasms which arise from or metastasize to the pituitary gland. The majority of pituitary neoplasms are adenomas, which are divided into non-secreting and secreting forms. Hormone producing forms are further classified by the type of hormone they secrete. Pituitary adenomas may also be characterized by their staining properties. Pituitary tumors may compress adjacent structures, including the hypothalamus, several cranial nerves, and the optic chiasm. Chiasmal compression may result in bitemporal hemianopsia. Pituitary Adenomas secreting GH or, less commonly, by extrapituitary disorders. Diagnosis involves neuroimaging Neuroimaging Non-invasive methods of visualizing the central nervous system, especially the brain, by various imaging modalities. Febrile Infant of the pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types and laboratory tests to evaluate the hypothalamic-pituitary axis Hypothalamic-pituitary axis Hypothalamic and Pituitary Hormones. Treatment depends on the operative status of the tumor Tumor Inflammation (if present), or a nonoperative treatment strategy may be utilized.
Last updated: Dec 15, 2025
For both acromegaly and gigantism, etiologies can be divided into 3 conceptual categories:
Regulation is a stepwise process:
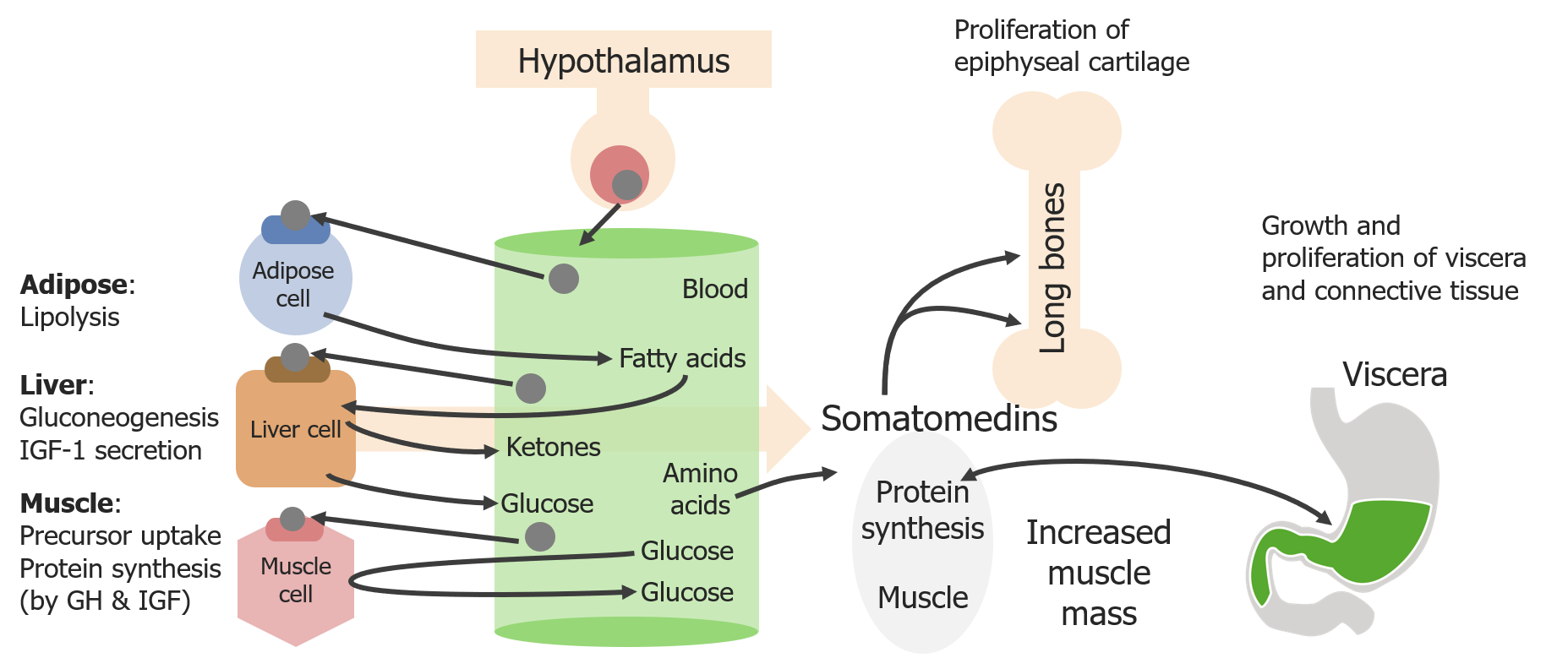
Schematic diagram of the direct and indirect effects of growth hormone (GH)
Image by Lecturio. License: CC BY-NC-SA 4.0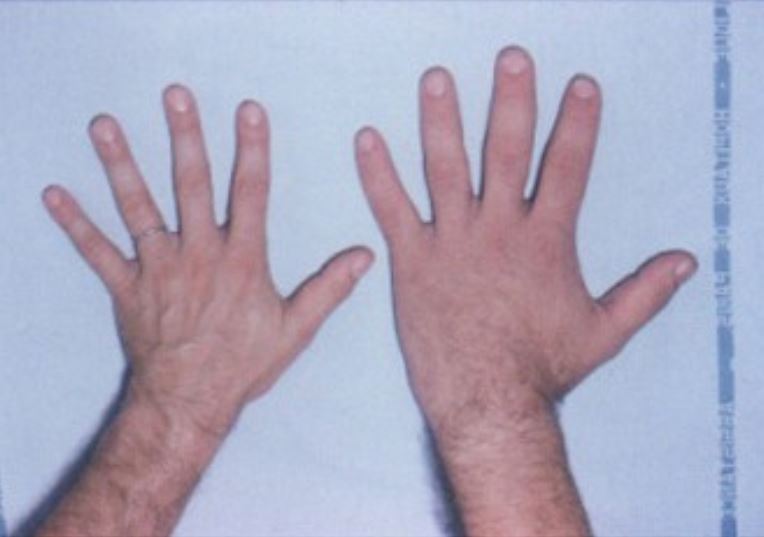
The hand of a healthy subject (left) next to the hand of a patient with acromegaly (right)
Image: “Acromegaly hands” by Philippe Chanson and Sylvie Salenave. License: CC BY 2.0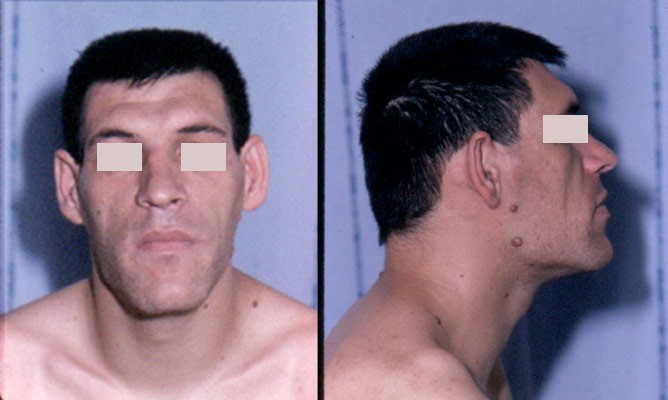
A patient with the typical facial features of acromegaly: enlarged nose, supraorbital bulges, prognathism, and a large head
Image: “Acromegaly facial features” by Philippe Chanson and Sylvie Salenave. License: CC BY 2.0Clinical presentation is typically the most important aspect of diagnosis.
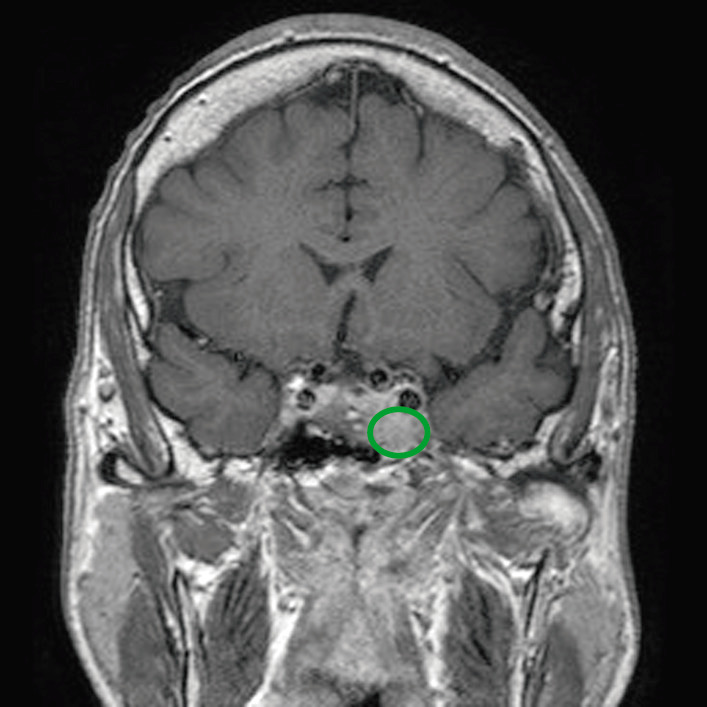
MRI of a patient with a pituitary adenoma (green circle) that is exerting mass effect on the surrounding structures
Image: “Before pasireotide therapy. MRI scan of pituitary – coronal view” by Rajesh Rajendran et al. License: CC BY 3.0, edited by Lecturio.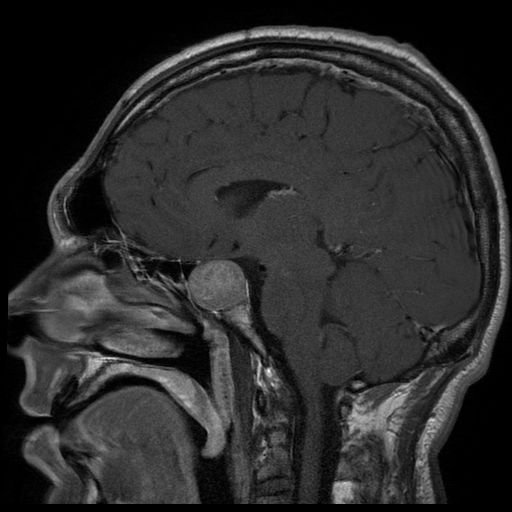
Magnetic resonance imaging (MRI) of a patient with a large pituitary adenoma, resulting in acromegaly
Image: “Acromegaly” by Elgee. License: CC BY 3.0