La acromegalia y el gigantismo son causados por una producción excesiva de la hormona de crecimiento (GH, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) por parte de la hipófisis. El gigantismo suele ser el resultado de un exceso de GH antes del cierre de la placa de crecimiento en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria niños. La acromegalia suele ser el resultado de un exceso de GH después del cierre de la placa de crecimiento. La estatura alta es un signo clínico observado en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum algunos pacientes con gigantismo. El aumento de la GH y el factor de crecimiento similar a la insulina-1 (IGF-1, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) son responsables de inducir el hipersomatotropismo. La acromegalia suele ser producida por tumores hipofisarios secretores de GH o, con menor frecuencia, por trastornos extrahipofisarios. El diagnóstico implica la imagenología neurológica de la hipófisis y pruebas de laboratorio para evaluar el eje hipotálamo-hipofisario. El tratamiento depende del estado quirúrgico del tumor Tumor Inflammation (si está presente), o se puede utilizar una estrategia terapéutica no quirúrgica.
Last updated: Dec 15, 2025
Tanto para la acromegalia como para el gigantismo, las etiologías pueden dividirse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 3 categorías conceptuales:
La regulación es un proceso gradual:
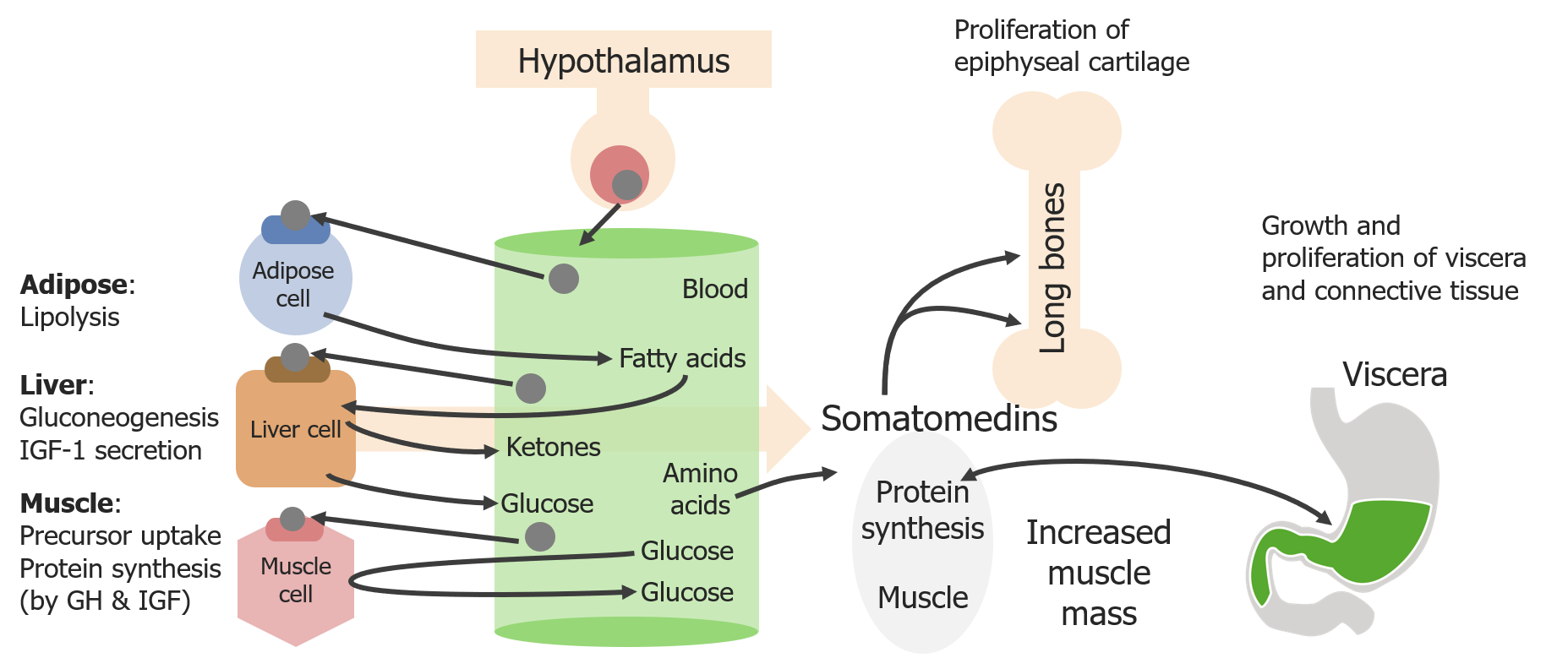
Diagrama esquemático de los efectos directos e indirectos de la hormona del crecimiento (GH)
Imagen por Lecturio. Licencia: CC BY-NC-SA 4.0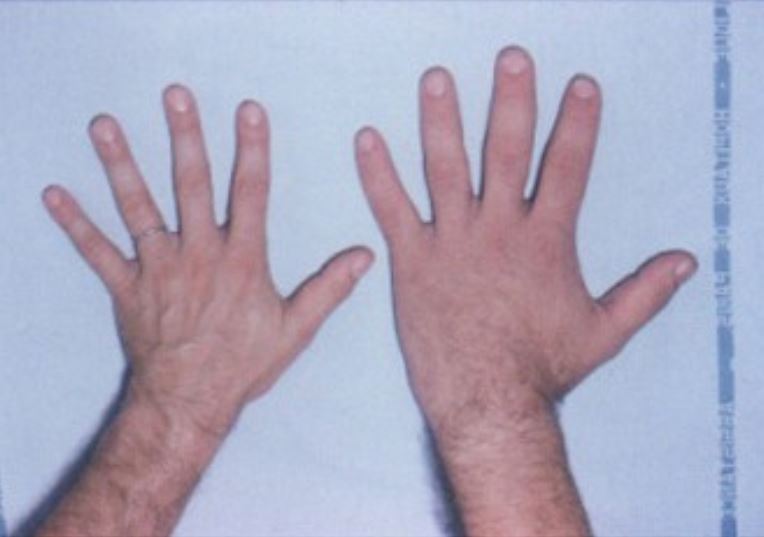
La mano de un sujeto sano (izquierda) junto a la mano de un paciente con acromegalia (derecha)
Imagen: “Acromegaly hands” por Philippe Chanson y Sylvie Salenave. Licencia: CC BY 2.0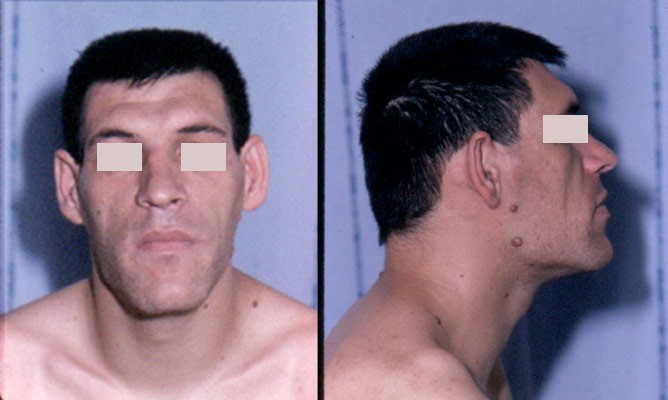
Un paciente con las características faciales típicas de la acromegalia: nariz agrandada, protuberancias supraorbitales, prognatismo y cabeza grande
Imagen: “Acromegaly facial features” por Philippe Chanson y Sylvie Salenave. Licencia: CC BY 2.0La presentación clínica suele ser el aspecto más importante del diagnóstico.
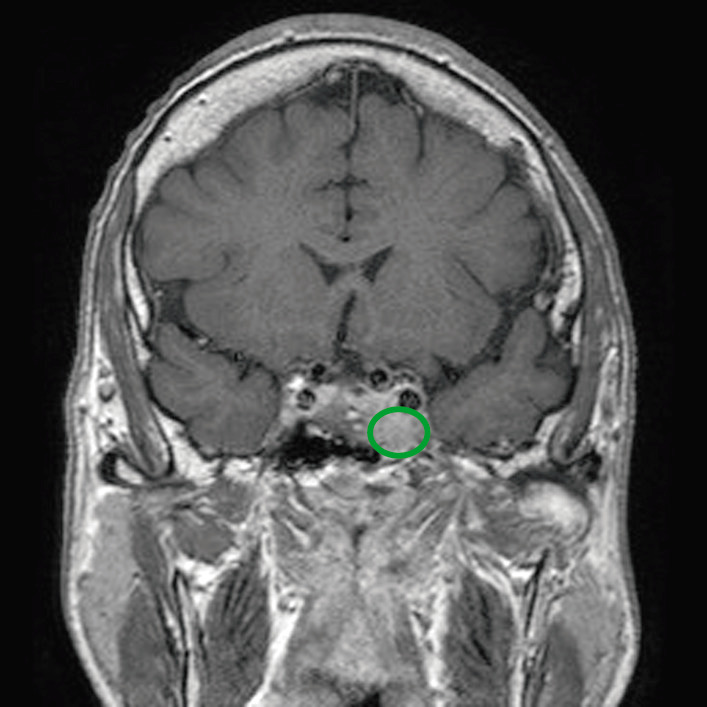
RM de un paciente con un adenoma hipofisario (círculo verde) que ejerce un efecto de masa sobre las estructuras circundantes
Imagen: “Before pasireotide therapy. MRI scan of pituitary – coronal view” por Rajesh Rajendran et al. Licencia: CC BY 3.0, editado por Lecturio.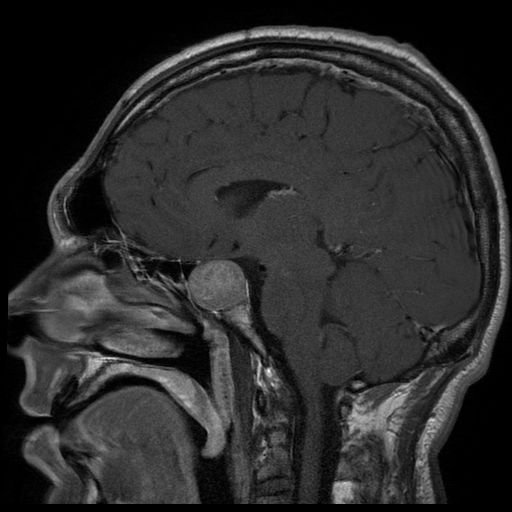
Resonancia magnética (RM) de un paciente con un gran adenoma hipofisario, que resultó en acromegalia
Imagen: “Acromegaly” por Elgee. Licencia: CC BY 3.0