


A acromegalia e o gigantismo são provocados por uma produção excessiva da hormona do crescimento (GH, pela sigla em inglês) pela hipófise. O gigantismo geralmente ocorre em crianças e resulta do excesso de GH antes do encerramento das placas de crescimento, nas crianças. A acromegalia geralmente resulta do excesso de GH após o encerramento das placas epifisárias. A alta estatura é um sinal clínico observado em alguns doentes com gigantismo. O aumento da GH e do fator de crescimento semelhante à insulina-1 (IGF-1, pela sigla em inglês) são responsáveis pela indução do hipersomatotropismo. A acromegalia é geralmente causada por tumores hipofisários que secretam GH ou, menos frequentemente, por distúrbios extra-hipofisários. O diagnóstico é realizado por neuroimagem da hipófise e com o auxílio de exames laboratoriais para avaliar o eixo hipotálamo-hipófise. O tratamento depende do estado cirúrgico do tumor Tumor Inflammation (se existir) ou pode ser adotada uma estratégia não cirúrgica.
Last updated: Dec 15, 2025
Tanto para a acromegalia como para o gigantismo, as etiologias podem ser divididas em 3 categorias conceptuais:
A regulação é um processo passo a passo:
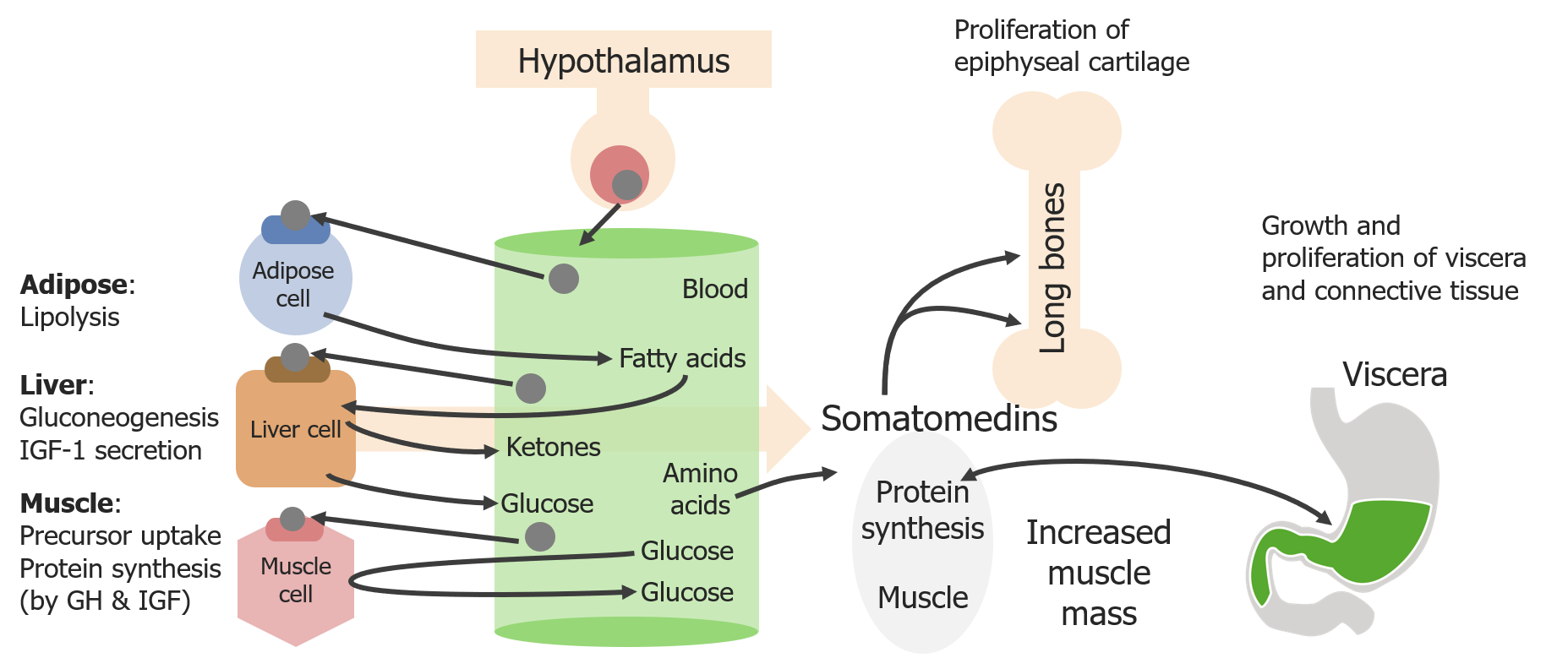
Diagrama esquemático dos efeitos diretos e indiretos da hormona do crescimento (GH)
Imagem por Lecturio. Licença: CC BY-NC-SA 4.0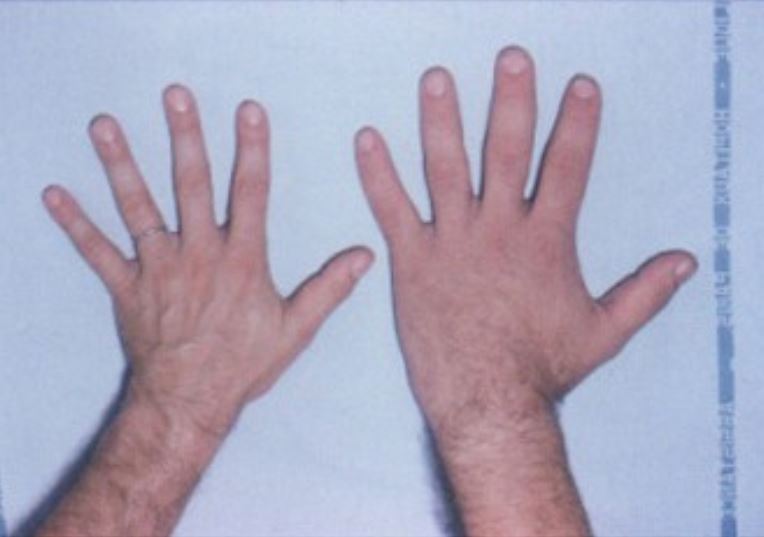
A mão de um indivíduo saudável (esquerda) ao lado da mão de um paciente com acromegalia (direita)
Imagem: “Acromegaly hands” por Philippe Chanson and Sylvie Salenave. Licença: CC BY 2.0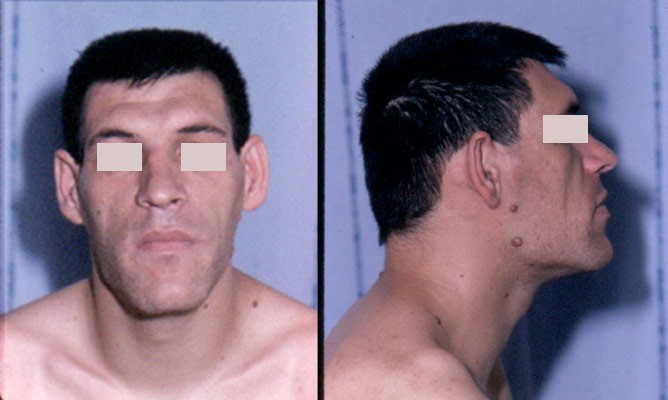
Paciente com as características faciais típicas de acromegalia: nariz aumentado, protuberâncias supraorbitárias, prognatismo e cabeça grande
Imagem: “Acromegaly facial features” por Philippe Chanson and Sylvie Salenave. Licença: CC BY 2.0A apresentação clínica é normalmente o ponto mais MAIS Androgen Insensitivity Syndrome importante do diagnóstico.
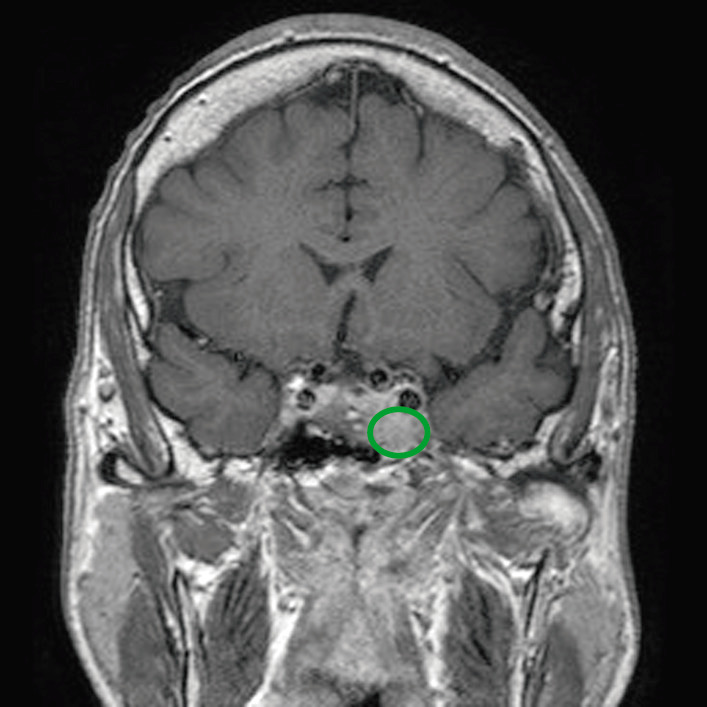
Ressonância magnética de paciente com adenoma hipofisário (círculo verde) a exercer efeito de massa nas estruturas adjacentes
Imagem: “Before pasireotide therapy. MRI scan of pituitary – coronal view” por Rajesh Rajendran et al. Licença: CC BY 3.0, editado por Lecturio.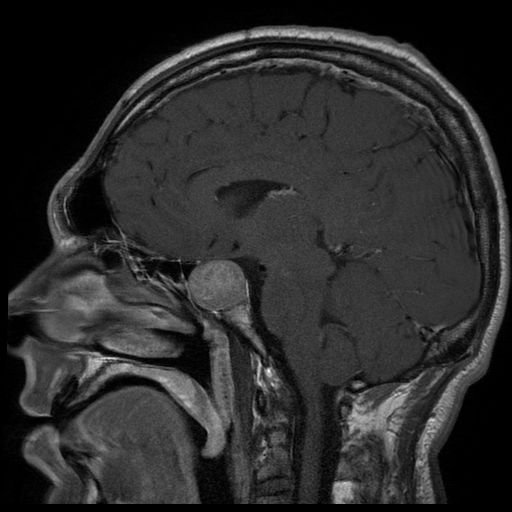
Imagem de ressonância magnética de paciente com adenoma hipofisário extenso a provocar acromegalia
Imagem: “Acromegaly” por Elgee. Licença: CC BY 3.0