Pituitary adenomas, also called pituitary neuroendocrine tumors or PitNETs in the 2022 WHO classification, are tumors that develop within the anterior lobe of the pituitary gland. While the WHO reclassified these as neuroendocrine tumors in 2022, the term “adenoma” remains widely used in clinical practice. They are classified by size (either micro- or macroadenomas) and by their ability to secrete hormones. Non-functioning or non-secretory adenomas do not secrete hormones but can compress surrounding pituitary tissue, leading to hypopituitarism. Secretory adenomas produce various hormones depending on the cell type from which they originated, leading to hyperpituitarism.
Last updated: Dec 15, 2025
General pathology
Classification by size
Classification by hormone production
| Hormone | Cell type | Target organ | Function | Increased | Decreased |
|---|---|---|---|---|---|
| ACTH | Corticotroph | Adrenal cortex Adrenal Cortex The outer layer of the adrenal gland. It is derived from mesoderm and comprised of three zones (outer zona glomerulosa, middle zona fasciculata, and inner zona reticularis) with each producing various steroids preferentially, such as aldosterone; hydrocortisone; dehydroepiandrosterone; and androstenedione. Adrenal cortex function is regulated by pituitary adrenocorticotropin. Adrenal Glands: Anatomy | Stimulate adrenocortical hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types |
|
Addison disease |
| Growth hormone | Somatotroph | Liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy and adipose tissue Adipose tissue Adipose tissue is a specialized type of connective tissue that has both structural and highly complex metabolic functions, including energy storage, glucose homeostasis, and a multitude of endocrine capabilities. There are three types of adipose tissue, white adipose tissue, brown adipose tissue, and beige or “brite” adipose tissue, which is a transitional form. Adipose Tissue: Histology | Stimulates protein synthesis Synthesis Polymerase Chain Reaction (PCR) and overall growth |
|
Dwarfism |
| Prolactin Prolactin A lactogenic hormone secreted by the adenohypophysis. It is a polypeptide of approximately 23 kd. Besides its major action on lactation, in some species prolactin exerts effects on reproduction, maternal behavior, fat metabolism, immunomodulation and osmoregulation. Breasts: Anatomy | Lactotroph | Mammary glands | Secretion Secretion Coagulation Studies of milk, female breast development | Hyperprolactinemia Hyperprolactinemia Hyperprolactinemia is defined as a condition of elevated levels of prolactin (PRL) hormone in the blood. The PRL hormone is secreted by the anterior pituitary gland and is responsible for breast development and lactation. The most common cause is PRL-secreting pituitary adenomas (prolactinomas). Hyperprolactinemia | |
| Thyroid- stimulating hormone | Thyrotroph | Thyroid Thyroid The thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck. Thyroid Gland: Anatomy gland | Stimulates synthesis Synthesis Polymerase Chain Reaction (PCR) and secretion Secretion Coagulation Studies of thyroid Thyroid The thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck. Thyroid Gland: Anatomy hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types | Hyperthyroidism Hyperthyroidism Hypersecretion of thyroid hormones from the thyroid gland. Elevated levels of thyroid hormones increase basal metabolic rate. Thyrotoxicosis and Hyperthyroidism | Hypothyroidism Hypothyroidism Hypothyroidism is a condition characterized by a deficiency of thyroid hormones. Iodine deficiency is the most common cause worldwide, but Hashimoto’s disease (autoimmune thyroiditis) is the leading cause in non-iodine-deficient regions. Hypothyroidism |
| Luteinizing hormone | Gonadotroph | Ovaries Ovaries Ovaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes). Ovaries: Anatomy and testes Testes Gonadal Hormones | Stimulates testosterone Testosterone A potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol. Androgens and Antiandrogens, ovulation Ovulation The discharge of an ovum from a rupturing follicle in the ovary. Menstrual Cycle, corpus luteum Corpus Luteum The yellow body derived from the ruptured ovarian follicle after ovulation. The process of corpus luteum formation, luteinization, is regulated by luteinizing hormone. Ovaries: Anatomy, estrogen Estrogen Compounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds. Ovaries: Anatomy and progesterone Progesterone The major progestational steroid that is secreted primarily by the corpus luteum and the placenta. Progesterone acts on the uterus, the mammary glands and the brain. It is required in embryo implantation; pregnancy maintenance, and the development of mammary tissue for milk production. Progesterone, converted from pregnenolone, also serves as an intermediate in the biosynthesis of gonadal steroid hormones and adrenal corticosteroids. Gonadal Hormones |
|
|
| Follicle- stimulating hormone | Gonadotroph | Ovaries Ovaries Ovaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes). Ovaries: Anatomy and testes Testes Gonadal Hormones | Sperm maturation, growth of follicles in ovaries Ovaries Ovaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes). Ovaries: Anatomy | Gonadal dysgenesis Gonadal dysgenesis A number of syndromes with defective gonadal developments such as streak gonads and dysgenetic testes or ovaries. The spectrum of gonadal and sexual abnormalities is reflected in their varied sex chromosome (sex chromosomes) constitution as shown by the karyotypes of 45, X monosomy (Turner syndrome); 46, XX (gonadal dysgenesis, 46xx); 46, XY (gonadal dysgenesis, 46, xy); and sex chromosome mosaicism. Their phenotypes range from female, through ambiguous, to male. This concept includes gonadal agenesis. Wilms Tumor | Hypogonadism Hypogonadism Hypogonadism is a condition characterized by reduced or no sex hormone production by the testes or ovaries. Hypogonadism can result from primary (hypergonadotropic) or secondary (hypogonadotropic) failure. Symptoms include infertility, increased risk of osteoporosis, erectile dysfunction, decreased libido, and regression (or absence) of secondary sexual characteristics. Hypogonadism |
| γ-melanocyte-stimulating hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types | Corticotroph | Melanocytes Melanocytes Mammalian pigment cells that produce melanins, pigments found mainly in the epidermis, but also in the eyes and the hair, by a process called melanogenesis. Coloration can be altered by the number of melanocytes or the amount of pigment produced and stored in the organelles called melanosomes. The large non-mammalian melanin-containing cells are called melanophores. Skin: Structure and Functions, endothelial cells, monocytes Monocytes Large, phagocytic mononuclear leukocytes produced in the vertebrate bone marrow and released into the blood; contain a large, oval or somewhat indented nucleus surrounded by voluminous cytoplasm and numerous organelles. Innate Immunity: Phagocytes and Antigen Presentation, and keratinocytes Keratinocytes Epidermal cells which synthesize keratin and undergo characteristic changes as they move upward from the basal layers of the epidermis to the cornified (horny) layer of the skin. Successive stages of differentiation of the keratinocytes forming the epidermal layers are basal cell, spinous or prickle cell, and the granular cell. Skin: Structure and Functions | Increase melanin Melanin Insoluble polymers of tyrosine derivatives found in and causing darkness in skin (skin pigmentation), hair, and feathers providing protection against sunburn induced by sunlight. Carotenes contribute yellow and red coloration. Seborrheic Keratosis synthesis Synthesis Polymerase Chain Reaction (PCR) in melanocytes Melanocytes Mammalian pigment cells that produce melanins, pigments found mainly in the epidermis, but also in the eyes and the hair, by a process called melanogenesis. Coloration can be altered by the number of melanocytes or the amount of pigment produced and stored in the organelles called melanosomes. The large non-mammalian melanin-containing cells are called melanophores. Skin: Structure and Functions | Melasma Melasma Melasma is a benign skin condition characterized by hyperpigmentation of sun-exposed regions due to excess melanin production and deposition. The condition mainly affects women during their reproductive years, particularly those with darker skin tones. Melasma, solar lentigo, and post- inflammatory hyper- pigmentation |
Microadenomas (< 10 mm)
Macroadenomas (> 10 mm)
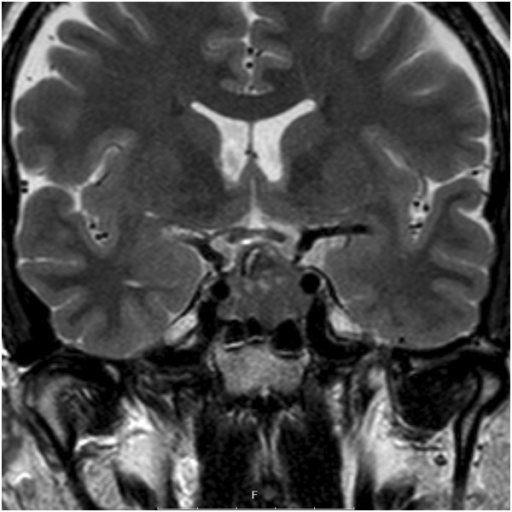
Coronal contrast MRI of the sella, showing a pituitary adenoma. No detectable invasion of the cavernous sinus.
Image: “Coronal T2-weighted magnetic resonance imaging of the sella, showing a pituitary adenoma” by Department of Clinical Sciences, Lund University, Lund S-221 85, Sweden. License: CC BY 4.0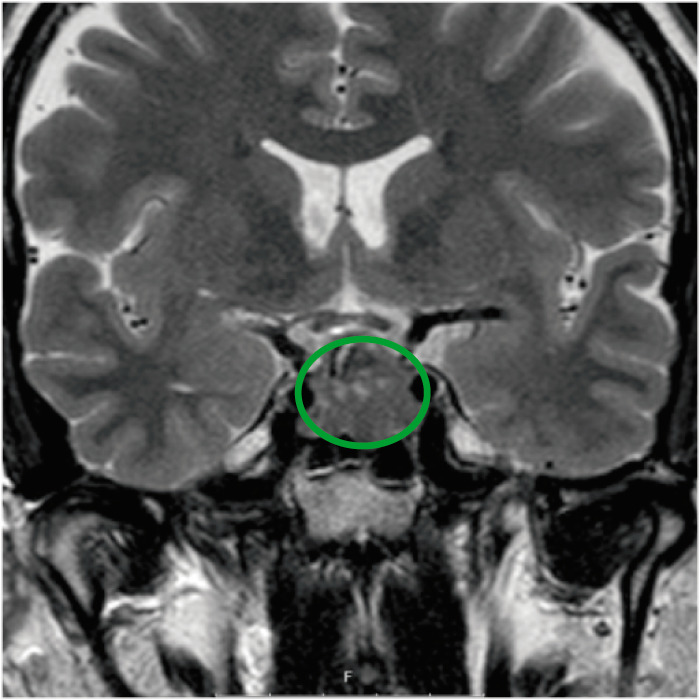
Coronal contrast MRI of the sella, showing a pituitary adenoma. No detectable invasion of the cavernous sinus.
Image: “Coronal T2-weighted magnetic resonance imaging of the sella, showing a pituitary adenoma” by Department of Clinical Sciences, Lund University, Lund S-221 85, Sweden. License: CC BY 4.0, edited by Lecturio.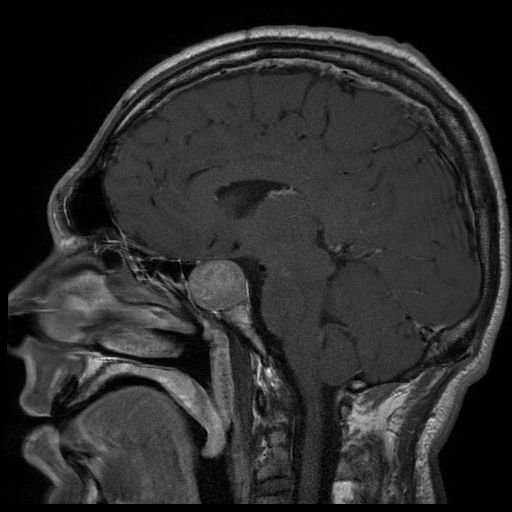
MRI of a patient presenting with acromegaly shows a large pituitary adenoma.
Image: “Tumeur hypophysaire” by Elgee. License: CC BY 3.0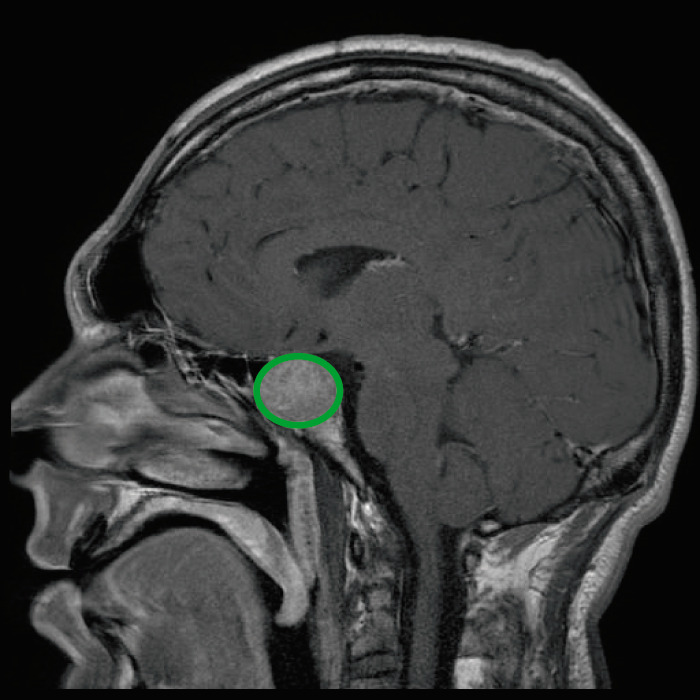
MRI of a patient presenting with acromegaly shows a large pituitary adenoma.
Image: “Tumeur hypophysaire” by Elgee. License: CC BY 3.0, edited by Lecturio.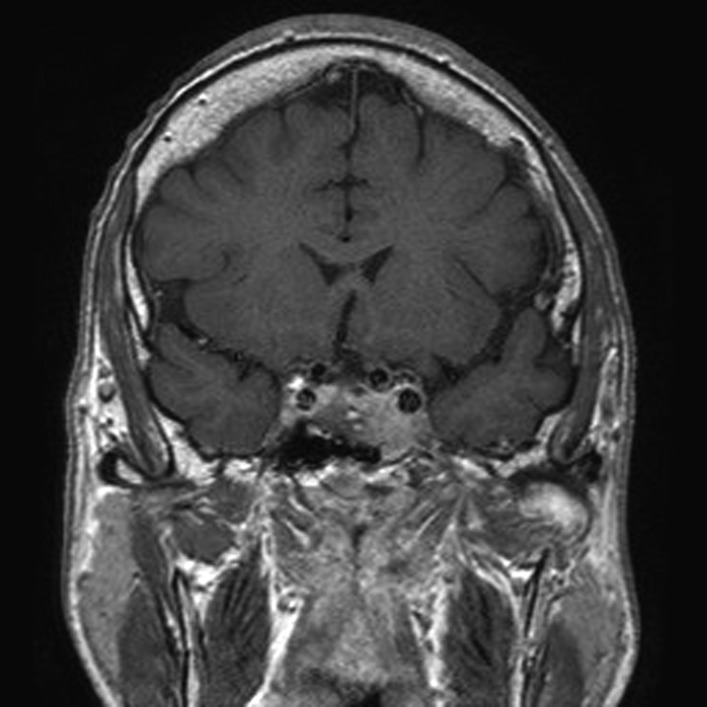
Contrast MRI scan of pituitary showing pituitary tumor with invasion of the cavernous sinus.
Image: “Before pasireotide therapy. MRI scan of pituitary – coronal view” by Rajesh Rajendran et al. License: CC BY 3.0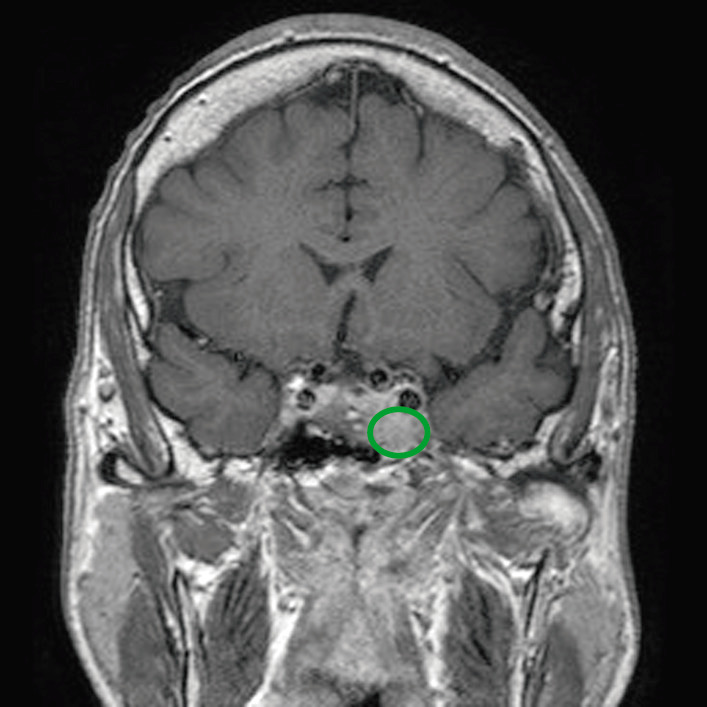
Contrast MRI scan of pituitary showing pituitary tumor with invasion of the cavernous sinus.
Image: “Before pasireotide therapy. MRI scan of pituitary – coronal view” by Rajesh Rajendran et al. License: CC BY 3.0, edited by Lecturio.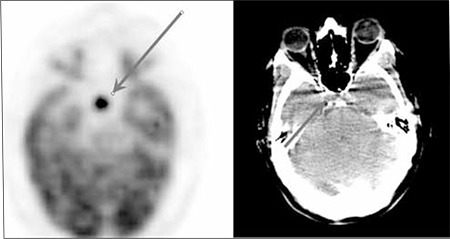
FDG-PET and CT axial image of an asymptomatic patient shows a focus of intense hypermetabolism in the sellar region, suggestive of a non-secretory pituitary adenoma.
Image: “FDG PET and CT axial image” by Sifa University, Nuclear Medicine, Izmir, Turkey. License: CC BY 2.5Treatment strategies depend on the tumor Tumor Inflammation cell type and size.
Medical therapy
Surgical resection
Transsphenoidal adenectomy (removal of the adenoma) or complete/partial hypophysectomy (removal of the pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types gland) is used when medical therapy fails. Endoscopic transsphenoidal adenectomy is now the preferred surgical approach, with complete adenoma removal achieved in 20-75% of cases depending on surgeon experience and center volume.
Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas
References