Cushing's syndrome or hypercortisolism is a disorder characterized by features resulting from chronic exposure to excess glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids. Cushing's syndrome may be exogenous, due to chronic glucocorticoid intake, or endogenous, due to increased adrenal secretion Secretion Coagulation Studies of cortisol Cortisol Glucocorticoids or adrenocorticotropic hormone Adrenocorticotropic hormone An anterior pituitary hormone that stimulates the adrenal cortex and its production of corticosteroids. Acth is a 39-amino acid polypeptide of which the n-terminal 24-amino acid segment is identical in all species and contains the adrenocorticotropic activity. Upon further tissue-specific processing, acth can yield alpha-msh and corticotropin-like intermediate lobe peptide (clip). Adrenal Hormones (ACTH) production from the pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types gland or ectopic sources. Exogenous or iatrogenic Iatrogenic Any adverse condition in a patient occurring as the result of treatment by a physician, surgeon, or other health professional, especially infections acquired by a patient during the course of treatment. Anterior Cord Syndrome hypercortisolism is the most common cause. Typical clinical features of hypercortisolism include central obesity Obesity Obesity is a condition associated with excess body weight, specifically with the deposition of excessive adipose tissue. Obesity is considered a global epidemic. Major influences come from the western diet and sedentary lifestyles, but the exact mechanisms likely include a mixture of genetic and environmental factors. Obesity, thin and bruisable skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions, abdominal striae, secondary hypertension Secondary hypertension Hypertension, hyperglycemia Hyperglycemia Abnormally high blood glucose level. Diabetes Mellitus, and proximal muscle weakness Proximal Muscle Weakness Lambert-Eaton Myasthenic Syndrome. The initial diagnostic approach is to establish hypercortisolism via urinary and salivary cortisol Cortisol Glucocorticoids tests along with low-dose dexamethasone Dexamethasone An anti-inflammatory 9-fluoro-glucocorticoid. Antiemetics suppression Suppression Defense Mechanisms test. Once the elevated cortisol Cortisol Glucocorticoids levels are confirmed, the etiology is determined based on ACTH levels, confirmatory biochemical tests, and subsequent imaging studies. Treatment options depend on the cause, and include surgery and medical therapy.
Last updated: Dec 15, 2025
Cushing’s syndrome or hypercortisolism is a disorder characterized by features resulting from chronic exposure to excess glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids.
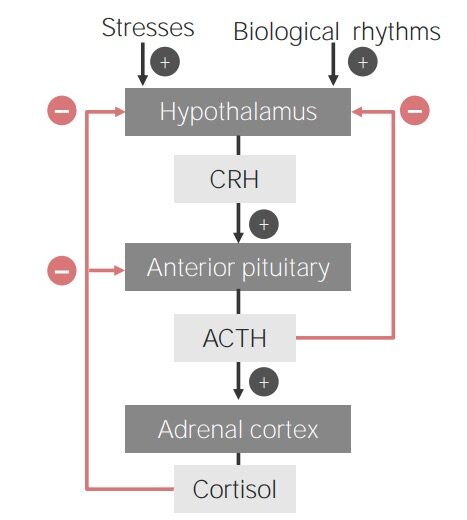
Flowchart showing the hypothalamic-pituitary-adrenal cortex axis
Image by Lecturio.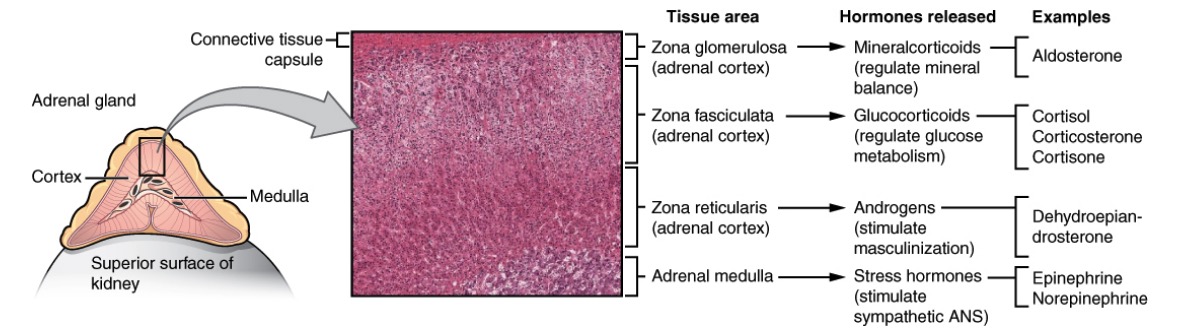
Anatomy, histology, and physiology of different zones of the adrenal gland:
The cortex has zona glomerulosa that produces mineralocorticoids (“salt”), zona fasciculata that produces glucocorticoids (“sugar”), and zona reticularis that produces androgens (“sex”). The adrenal medulla produces epinephrine and norepinephrine (“stress”).
| Skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions |
|
|---|---|
| Body fat | Moon facies, central obesity Obesity Obesity is a condition associated with excess body weight, specifically with the deposition of excessive adipose tissue. Obesity is considered a global epidemic. Major influences come from the western diet and sedentary lifestyles, but the exact mechanisms likely include a mixture of genetic and environmental factors. Obesity, buffalo hump (dorsocervical fat pad) |
| Bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types | Short stature (decreased linear growth in children), osteopenia Osteopenia Osteoporosis, osteoporosis Osteoporosis Osteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis, ↑ risk of avascular necrosis Avascular Necrosis Hip Fractures |
| Muscle | Proximal myopathy Myopathy Dermatomyositis, weakness |
| Metabolism | Elevated glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance/ diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus, dyslipidemia |
| Reproductive | Amenorrhea Amenorrhea Absence of menstruation. Congenital Malformations of the Female Reproductive System in women, decreased libido |
| Cardiovascular | Atherosclerosis Atherosclerosis Atherosclerosis is a common form of arterial disease in which lipid deposition forms a plaque in the blood vessel walls. Atherosclerosis is an incurable disease, for which there are clearly defined risk factors that often can be reduced through a change in lifestyle and behavior of the patient. Atherosclerosis, hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, edema Edema Edema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema |
| GI | Ulcers |
| Neuropsychiatric | Irritability, depression, emotional lability, sleep Sleep A readily reversible suspension of sensorimotor interaction with the environment, usually associated with recumbency and immobility. Physiology of Sleep disturbance, psychosis |
| Ophthalmologic | Cataracts ( steroids Steroids A group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus. Benign Liver Tumors affect gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics transcription Transcription Transcription of genetic information is the first step in gene expression. Transcription is the process by which DNA is used as a template to make mRNA. This process is divided into 3 stages: initiation, elongation, and termination. Stages of Transcription of lens Lens A transparent, biconvex structure of the eye, enclosed in a capsule and situated behind the iris and in front of the vitreous humor (vitreous body). It is slightly overlapped at its margin by the ciliary processes. Adaptation by the ciliary body is crucial for ocular accommodation. Eye: Anatomy epithelial cells) |
| Immune system Immune system The body’s defense mechanism against foreign organisms or substances and deviant native cells. It includes the humoral immune response and the cell-mediated response and consists of a complex of interrelated cellular, molecular, and genetic components. Primary Lymphatic Organs | Increased WBC count, increased susceptibility to infection |
To recall the most common clinical features of Cushing’s syndrome, remember “CUSHINGOID”:
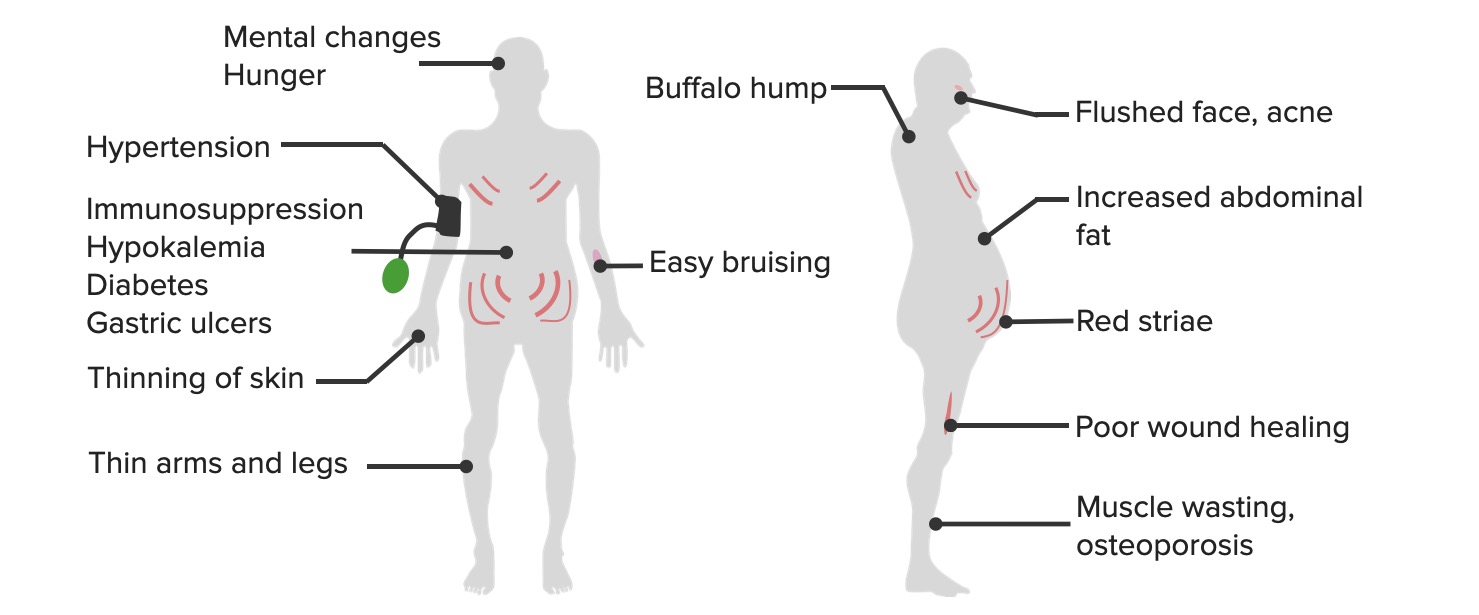
The common clinical manifestations of hypercortisolism
Image by Lecturio.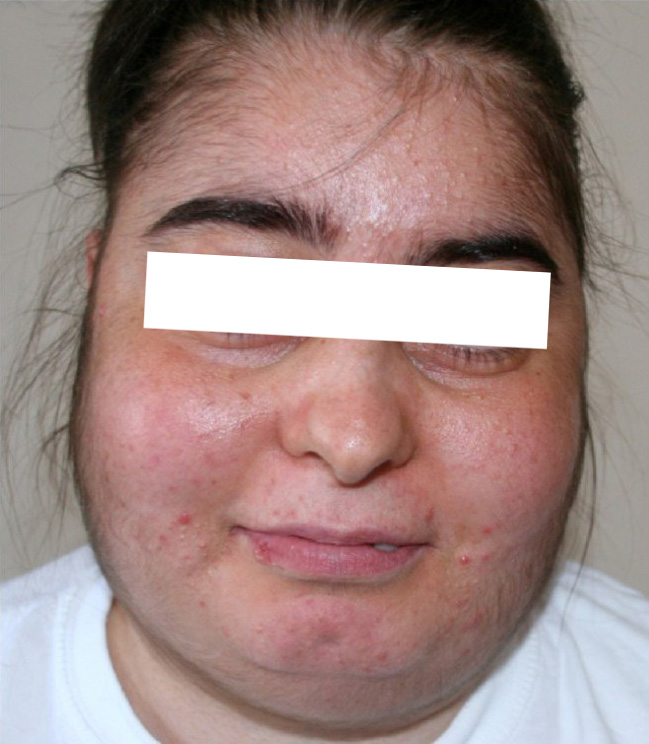
Features of Cushing’s syndrome: woman presenting with hirsutism, acne, and moon facies
Image: “Patient’s facial appearance” by Ozlem Celik et al. License: CC BY 2.5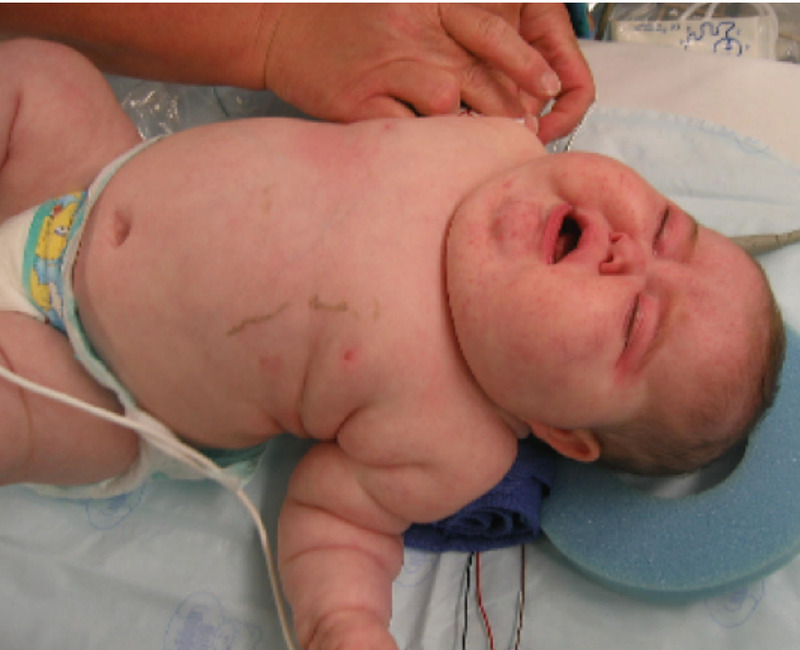
Infant with Cushing’s syndrome:
Features noted include moon facies, plethora, acne, central obesity, and poor muscle tone.
Clinical features increase suspicion of diagnosis:
Exclude use of exogenous glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids:
Initial tests for hypercortisolism:
Abnormal results prompt additional evaluation:
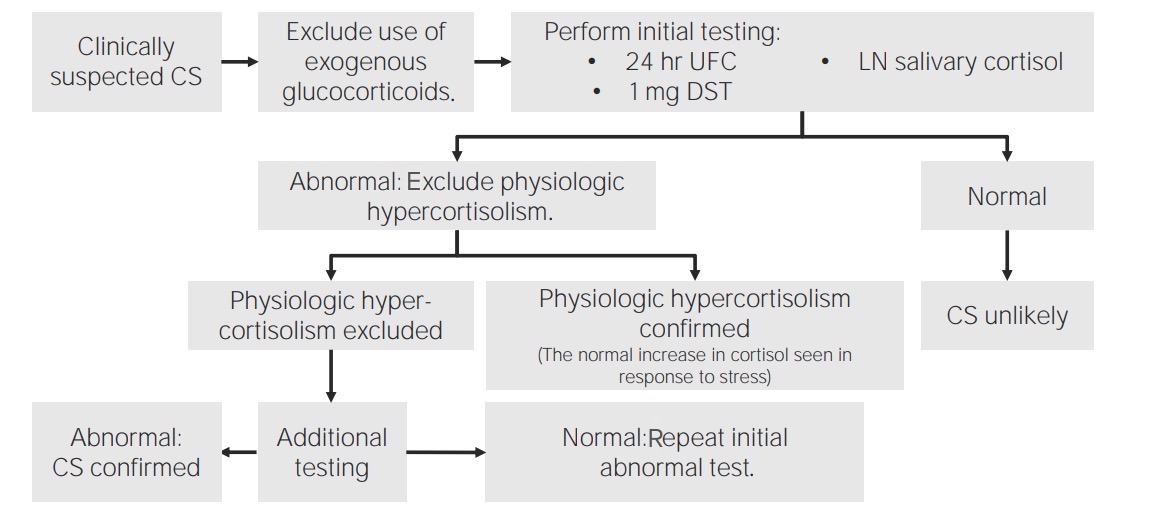
Algorithm to diagnose hypercortisolism or Cushing’s syndrome (CS):
In patients clinically suspected with CS, the 1st step is to rule out the exogenous use of glucocorticoids. Once excluded, initial tests to check for elevated cortisol levels include late-night (LN) salivary cortisol, 24-hour urinary free cortisol (UFC), and 1 mg dexamethasone suppression test (DST). Once elevated cortisol levels are confirmed, evaluate whether the elevation is from physiological hypercortisolism. Once physiologic causes are excluded, proceed with additional testing to identify the etiology (which can be primary or secondary).
After confirming elevated cortisol Cortisol Glucocorticoids levels, determining the etiology starts with ACTH.
| ACTH level | Etiology and additional tests | Identifying the cause |
|---|---|---|
| ↓ ACTH: ACTH-independent | Adrenal gland (possible adenoma, carcinoma, hyperplasia Hyperplasia An increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells. Cellular Adaptation) | Obtain CT scan/MRI of the adrenal gland. |
| ↑ or intermediate ACTH: likely ACTH-dependent | Cushing’s disease:
|
Obtain pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types MRI. |
Ectopic ACTH syndrome (such as
lung cancer
Lung cancer
Lung cancer is the malignant transformation of lung tissue and the leading cause of cancer-related deaths. The majority of cases are associated with long-term smoking. The disease is generally classified histologically as either small cell lung cancer or non-small cell lung cancer. Symptoms include cough, dyspnea, weight loss, and chest discomfort.
Lung Cancer, carcinoid):
|
Workup for
malignancy
Malignancy
Hemothorax:
|