Adrenal insufficiency (AI) is the inadequate production of adrenocortical hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types: glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids, mineralocorticoids Mineralocorticoids Mineralocorticoids are a drug class within the corticosteroid family and fludrocortisone is the primary medication within this class. Fludrocortisone is a fluorinated analog of cortisone. The fluorine moiety protects the drug from isoenzyme inactivation in the kidney, allowing it to exert its mineralocorticoid effect. Mineralocorticoids, and adrenal androgens Androgens Androgens are naturally occurring steroid hormones responsible for development and maintenance of the male sex characteristics, including penile, scrotal, and clitoral growth, development of sexual hair, deepening of the voice, and musculoskeletal growth. Androgens and Antiandrogens. Primary AI, also called Addison’s disease, is caused by adrenal gland disorder (autoimmune disease, infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, and malignancy Malignancy Hemothorax, among others). Adrenal insufficiency can also occur because of decreased production of adrenocorticotropic hormone Adrenocorticotropic hormone An anterior pituitary hormone that stimulates the adrenal cortex and its production of corticosteroids. Acth is a 39-amino acid polypeptide of which the n-terminal 24-amino acid segment is identical in all species and contains the adrenocorticotropic activity. Upon further tissue-specific processing, acth can yield alpha-msh and corticotropin-like intermediate lobe peptide (clip). Adrenal Hormones (ACTH) from disease in the pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types gland (secondary) or hypothalamic disorders and prolonged glucocorticoid therapy (tertiary). Diagnosis is by demonstrating hypocortisolism (via cortisol Cortisol Glucocorticoids and ACTH levels and ACTH-stimulation test) and determining the etiology (adrenal autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques, imaging). Glucocorticoid replacement is needed in all forms of AI. Additionally, in primary AI, mineralocorticoid is given to prevent volume depletion Volume depletion Volume status is a balance between water and solutes, the majority of which is Na. Volume depletion refers to a loss of both water and Na, whereas dehydration refers only to a loss of water. Volume depletion can be caused by GI losses, renal losses, bleeding, poor oral Na intake, or third spacing of fluids. Volume Depletion and Dehydration, salt loss, and hyperkalemia Hyperkalemia Hyperkalemia is defined as a serum potassium (K+) concentration >5.2 mEq/L. Homeostatic mechanisms maintain the serum K+ concentration between 3.5 and 5.2 mEq/L, despite marked variation in dietary intake. Hyperkalemia can be due to a variety of causes, which include transcellular shifts, tissue breakdown, inadequate renal excretion, and drugs. Hyperkalemia. Adrenal crisis is a medical emergency; management requires prompt IV hydration Iv Hydration Crush Syndrome and administration of IV glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids without waiting for initial hormone results.
Last updated: May 17, 2024
Adrenal insufficiency (AI) is the deficiency in adrenal production of glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids, adrenal androgens Androgens Androgens are naturally occurring steroid hormones responsible for development and maintenance of the male sex characteristics, including penile, scrotal, and clitoral growth, development of sexual hair, deepening of the voice, and musculoskeletal growth. Androgens and Antiandrogens, and mineralocorticoids Mineralocorticoids Mineralocorticoids are a drug class within the corticosteroid family and fludrocortisone is the primary medication within this class. Fludrocortisone is a fluorinated analog of cortisone. The fluorine moiety protects the drug from isoenzyme inactivation in the kidney, allowing it to exert its mineralocorticoid effect. Mineralocorticoids.

Adrenal glands:
Small, triangular glands that sit on top of the kidneys (suprarenal glands)
Consequence of dysfunction of the hypothalamus Hypothalamus The hypothalamus is a collection of various nuclei within the diencephalon in the center of the brain. The hypothalamus plays a vital role in endocrine regulation as the primary regulator of the pituitary gland, and it is the major point of integration between the central nervous and endocrine systems. Hypothalamus and pituitary Pituitary A small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum. Hormones: Overview and Types gland:
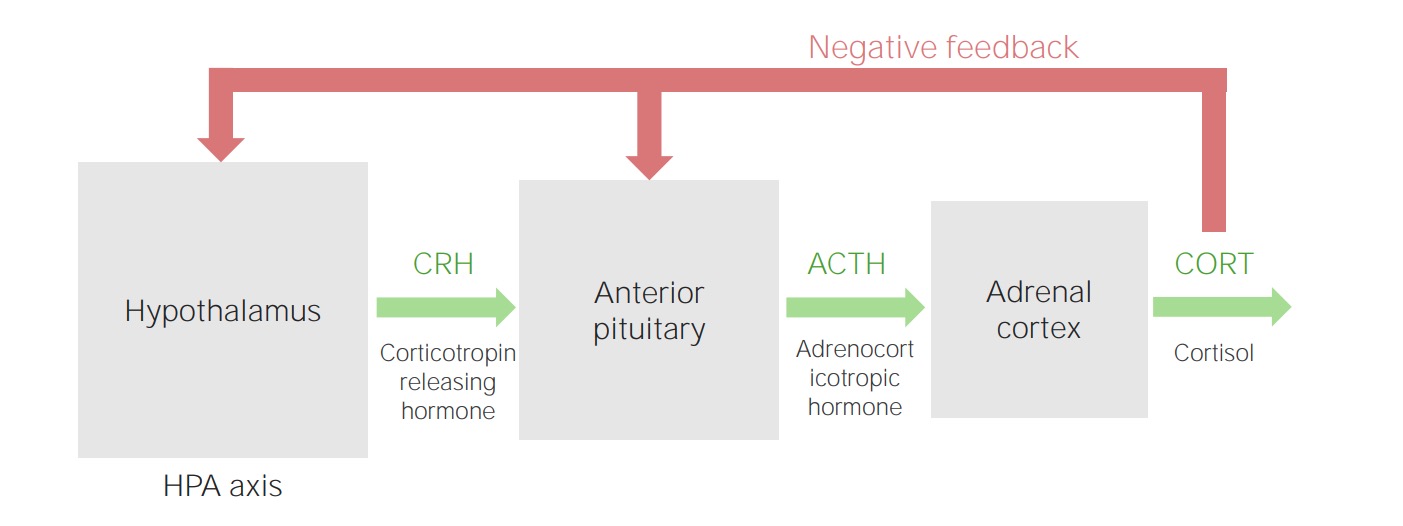
Hypothalamic–pituitary–adrenal cortex axis
Image by Lecturio.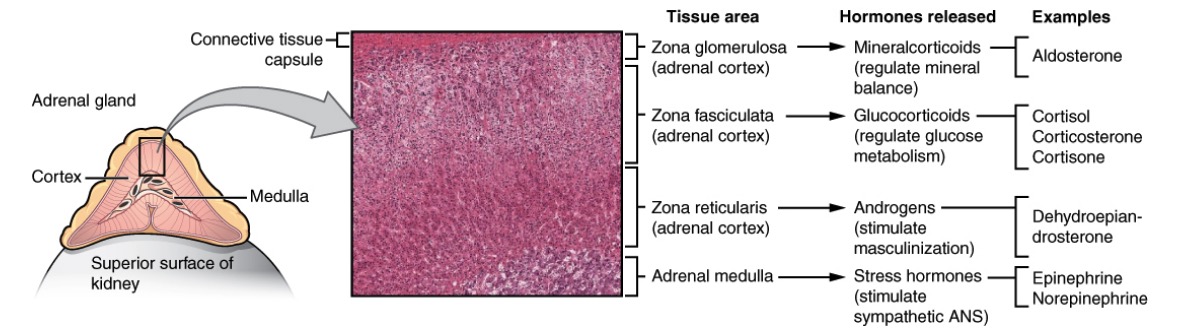
Anatomy, histology, and physiology of different zones of the adrenal gland:
The cortex has zona glomerulosa that produces mineralocorticoids (salt), zona fasciculata that produces glucocorticoids (sugar), and zona reticularis that produces androgens (sex). The adrenal medulla produces epinephrine and norepinephrine (stress).
Primary AI:
Secondary AI:
Tertiary AI:

Normal adrenal function
Image by Lecturio.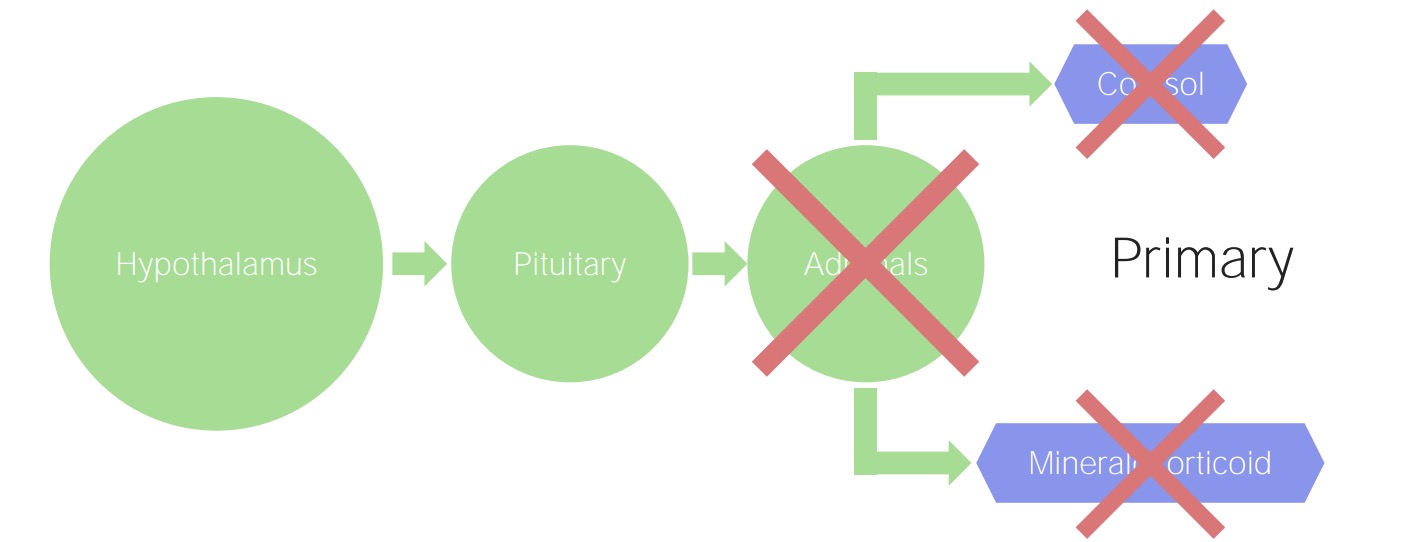
Primary adrenal insufficiency:
The adrenals are damaged, regardless of the etiology. Consequently, there is a decline in the production of cortisol and mineralocorticoids.
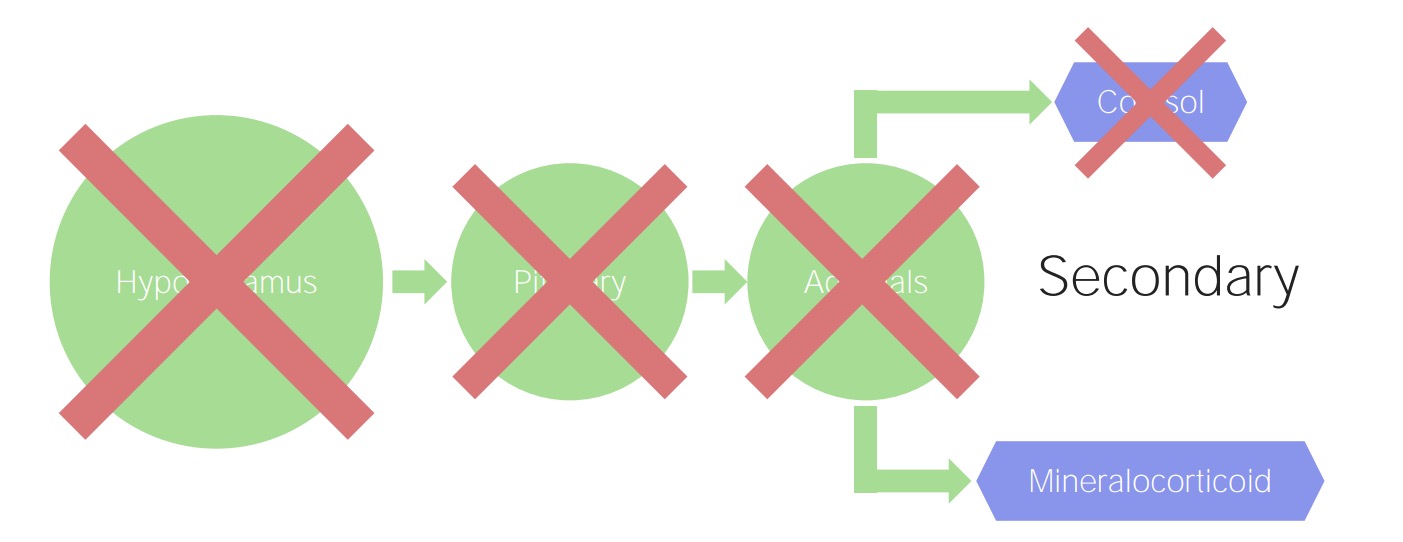
Secondary and tertiary adrenal insufficiency:
A pituitary (secondary) or hypothalamic (tertiary) problem results in decreased stimulation of cortisol. However, mineralocorticoid production is regulated by the renin–angiotensin–aldosterone system, so this adrenal function remains preserved.

Manifestation of primary adrenal insufficiency:
Patient presents initially with hyperpigmentation (left). Note the change in skin tone after treatment is administered (right).
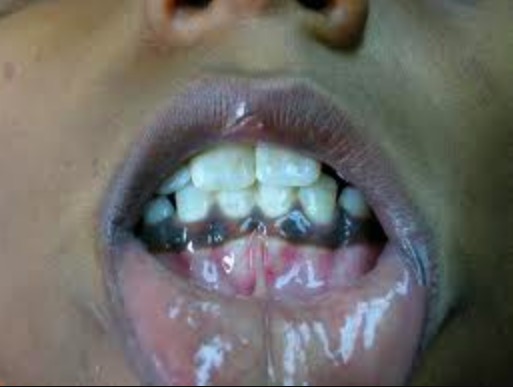
Hyperpigmentation:
Patient with adrenal insufficiency presents with hyperpigmentation of the oral mucosa.
Adrenal crisis is the acute decompensation of adrenal function that can be triggered by another disease, surgery, stress, or increased glucocorticoid inactivation ( hyperthyroidism Hyperthyroidism Hypersecretion of thyroid hormones from the thyroid gland. Elevated levels of thyroid hormones increase basal metabolic rate. Thyrotoxicosis and Hyperthyroidism).
Presentation:
| Primary AI | Secondary or tertiary AI | |
|---|---|---|
| Morning cortisol Cortisol Glucocorticoids | ↓ | ↓ |
| ACTH | ↑ | ↓ or normal |
| Renin Renin A highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system. Renal Sodium and Water Regulation concentration | ↑ | Normal |
| Aldosterone Aldosterone A hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium. Hyperkalemia | ↓ | Normal |
| Electrolytes Electrolytes Electrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions. Electrolytes |
|
|
| ACTH-stimulation test | No increase in cortisol Cortisol Glucocorticoids | Increase in cortisol Cortisol Glucocorticoids* |
Performed when initial tests are indeterminate
In suspected primary AI:
In suspected secondary/tertiary AI:
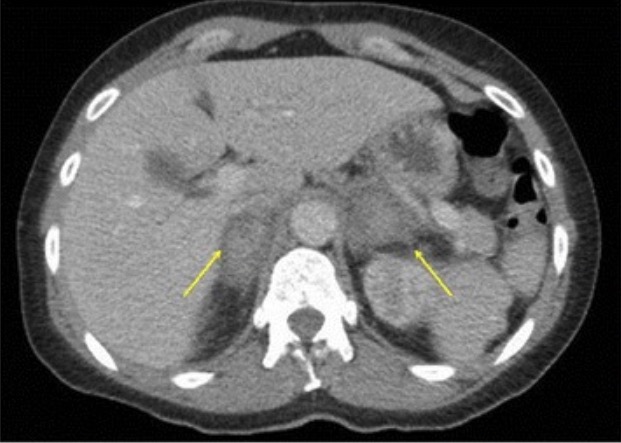
CT of adrenal hemorrhage:
Contrast imaging of the abdomen and pelvis showing bilateral adrenal hemorrhage (yellow arrows) in a patient presenting with sepsis
Primary AI:
Secondary and tertiary AI: