{kind=link}



A insuficiência suprarrenal (IA, pela sigla em inglês) é a produção inadequada de hormonas adrenocorticais: glicocorticoides, mineralocorticoides e androgénios adrenais. A IA primária, também denominada doença de Addison, é causada por um distúrbio da glândula adrenal (doença autoimune, infeções e malignidade, entre outras). A insuficiência suprarrenal também pode ocorrer devido à diminuição da produção da hormona adrenocorticotrófica (ACTH, pela sigla em inglês) por doença na glândula pituitária (secundária) ou distúrbios hipotalâmicos e por terapêutica prolongada com glicocorticoides (terciária). O diagnóstico é feito pela demonstração de hipocortisolismo (através da medição dos níveis de cortisol Cortisol Glucocorticoids e ACTH e do teste de estimulação da ACTH) e determinação da etiologia (autoanticorpos suprarrenais, imagem). A reposição de glicocorticoides é necessária em todas as formas de IA. Além disso, na IA primária, são administrados mineralocorticoides para prevenir a depleção de volume, a perda de sal e a hipercalemia. A crise suprarrenal é uma emergência médica; o tratamento requer hidratação IV imediata e administração de glicocorticoides IV, sem esperar pelos doseamentos hormonais iniciais.
Last updated: Jun 13, 2022
A insuficiência suprarrenal (IA, pela sigla em inglês) é a deficiência na produção suprarrenal de glicocorticoides, androgénios suprarrenal e mineralocorticoides.

Glândulas suprarrenais (ou adrenais):
Glândulas pequenas, triangulares que se situam sobre os rins (suprarenais)
Consequências da disfunção do hipotálamo e da hipófise:
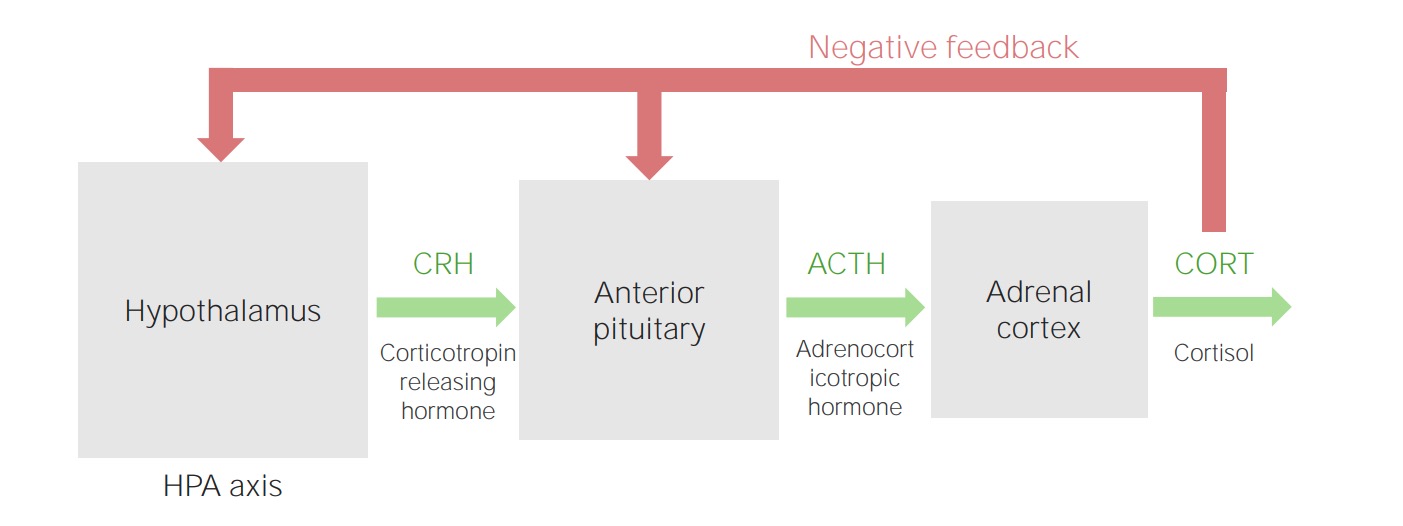
Eixo hipotalâmico-hipofisário-córtex suprarrenal
Imagem por Lecturio.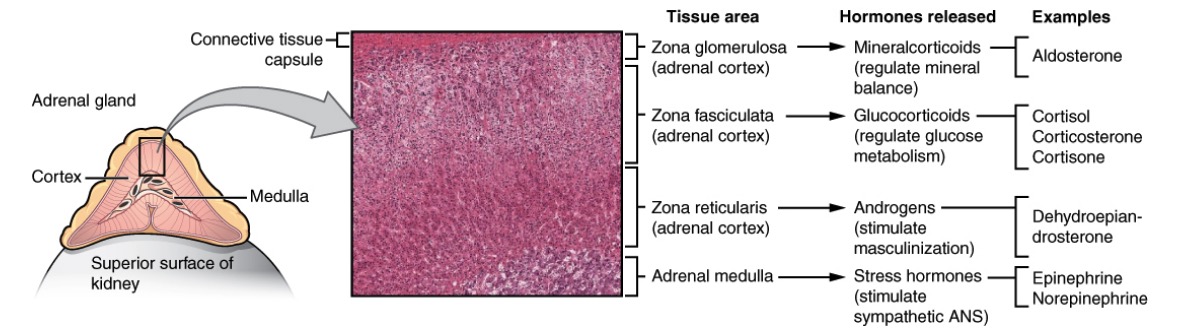
Anatomia, histologia e fisiologia de diferentes zonas da glândula suprarrenal:
O córtex tem a zona glomerulosa que produz mineralocorticoides (sal), a zona fasciculada que produz glicocorticoides (açúcar) e a zona reticular que produz androgénios (sexo). A medula suprarrenal produz epinefrina e norepinefrina (stress).
IA primária:
IA secundária:
IA terciária:

Função adrenal normal
Imagem por Lecturio.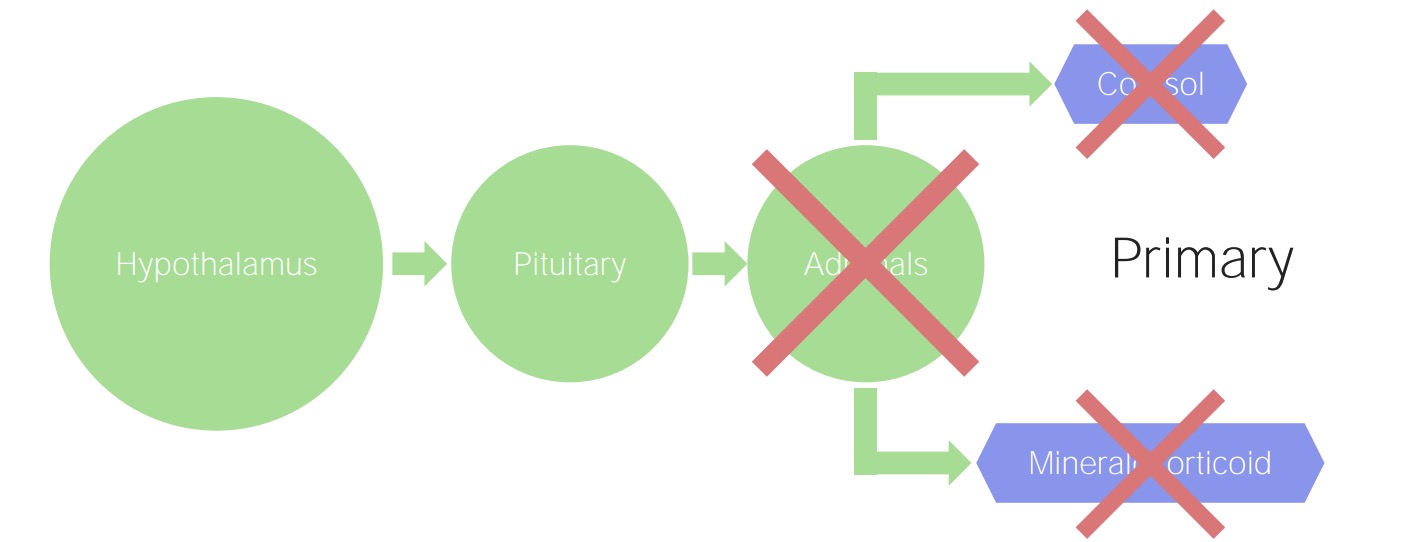
Insuficiência suprarrenal primária:
As suprarrenais estão danificadas, independentemente da etiologia. Consequentemente, há um declínio na produção de cortisol e mineralocorticoides.
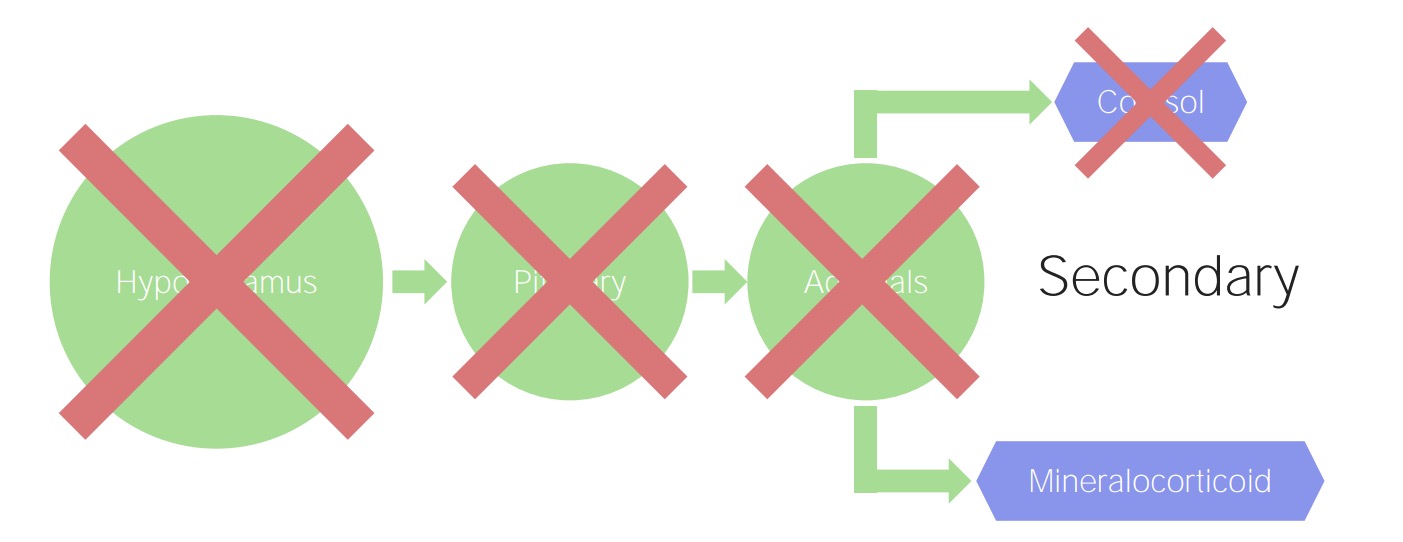
Insuficiência suprarrenal secundária e terciária:
Um problema hipofisário (secundário) ou hipotalâmico (terciário) resulta na diminuição da estimulação do cortisol. No entanto, a produção de mineralocorticoides é regulada pelo sistema renina-angiotensina-aldosterona, de forma que essa função da suprarrenal permanece preservada.

Manifestação de insuficiência suprarrenal primária:
O dooente apresenta inicialmente hiperpigmentação (esquerda). Observar a alteração no tom da pele após a administração do tratamento (à direita).
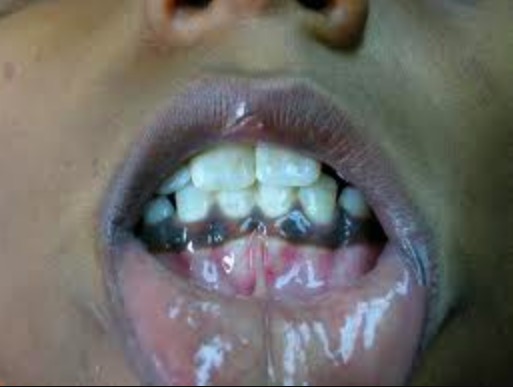
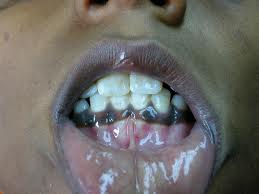
Hiperpigmentação:
Doente com insuficiência suprarrenal que apresenta hiperpigmentação da mucosa oral.
A crise adrenal é a descompensação aguda da função da suprarrenal, que pode ser desencadeada por outra doença, cirurgia, stress ou por aumento do processo de inativação dos glicocorticoides (hipertiroidismo).
Apresentação Clínica:
| IA primária | IA secundária ou terciária | |
|---|---|---|
| Cortisol Cortisol Glucocorticoids matinal | ↓ | ↓ |
| ACTH | ↑ | ↓ ou normal |
| Concentração de renina | ↑ | Normal |
| Aldosterona | ↓ | Normal |
| Eletrólitos |
|
|
| Teste de estimulação da ACTH | Sem aumento do cortisol Cortisol Glucocorticoids | Aumento do cortisol Cortisol Glucocorticoids* |
Realizados quando os exames iniciais apresentam resultados indeterminados
Na suspeita de IA primária:
Na suspeita de IA secundária/terciária:
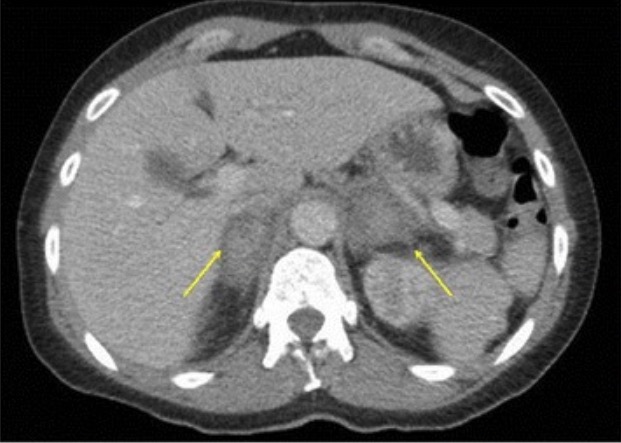
TC com hemorragia suprarrenal:
Imagem contrastada do abdómen e da pelve a demonstrar hemorragia suprarrenal bilateral (setas amarelas) num doente com sépsis
IA primária:
IA secundária e terciária:
