Spinal muscular atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation (SMA) is a spectrum of autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance syndromes characterized by progressive proximal muscle weakness Proximal Muscle Weakness Lambert-Eaton Myasthenic Syndrome and atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation, possibly due to degeneration of the anterior horn Anterior horn One of three central columns of the spinal cord. It is composed of gray matter spinal laminae VIII and ix. Brown-Séquard Syndrome cells in the spinal cord Spinal cord The spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy and motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology nuclei in the lower brainstem. There are 5 clinical types of SMA, each with a distinctive clinical presentation unified by motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology weakness. The earlier presentations are associated with more severe motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology weakness affecting a child’s ability to reach the developmental milestones Developmental milestones Developmental milestones are the skills or abilities that most children are able to perform when they reach a certain age. Understanding the appropriate milestones and at what age they are reached helps clinicians identify symptoms of delayed development. Developmental milestones are divided into 5 important domains: gross motor, fine motor, language, social, and cognitive. Developmental Milestones and Normal Growth of sitting or walking. In the more severe types, breathing and swallowing Swallowing The act of taking solids and liquids into the gastrointestinal tract through the mouth and throat. Gastrointestinal Motility may also become difficult as the disease progresses. The prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas of SMA is poor. In the less severe types, adults have a normal lifespan. The initial diagnosis is made clinically and confirmed using genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Management is mostly supportive although novel therapies are being developed. The prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas depends on the clinical type.
Last updated: Dec 15, 2025
Spinal muscular atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation (SMA) is a spectrum of autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance syndromes characterized by progressive proximal muscle weakness Proximal Muscle Weakness Lambert-Eaton Myasthenic Syndrome and atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation, possibly due to degeneration of the anterior horn Anterior horn One of three central columns of the spinal cord. It is composed of gray matter spinal laminae VIII and ix. Brown-Séquard Syndrome cells in the spinal cord Spinal cord The spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy and motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology nuclei in the lower brainstem.
Spinal muscular atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation presents as a lower motor neuron Lower Motor Neuron Motor Neuron Lesions lesion syndrome with muscle weakness, which is more severe in types 0, 1, and 2.
Spinal muscular atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation should be suspected in any infant with unexplained weakness or hypotonia Hypotonia Duchenne Muscular Dystrophy. Definitive diagnosis requires genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies.
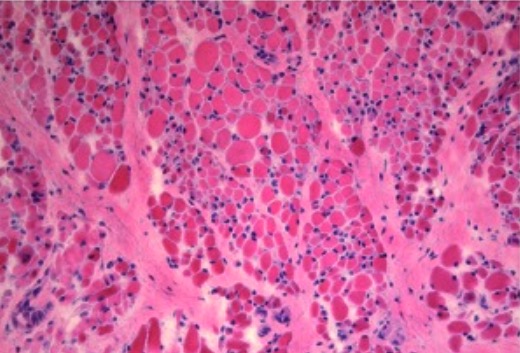
Muscle biopsy at 10 months of age (H&E, ×200) with marked perimysial and endomysial fibrosis and fatty infiltration with groups of small fibers and scattered hypertrophic fibers
Image: “Muscle biopsy in SMA” by . License: CC BY 2.5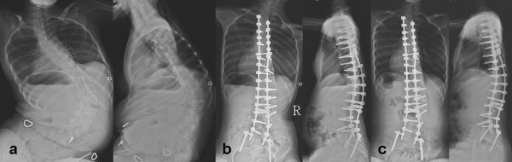
Images of a 13-year-old girl with spinal muscle atrophy (SMA):
a: preoperative anteroposterior (AP) and lateral radiogram of the spine
b: postoperative AP and lateral radiogram of the spine
c: follow-up AP and lateral radiograph of the spine
Current management is mostly supportive and multidisciplinary (including palliative care).
Gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics therapy: 3 agents are now available in the US.