


Atrofia muscular espinhal (AME) é um espectro de síndromes autossómicas recessivas caracterizadas por fraqueza e atrofia muscular proximal progressiva, possivelmente devido à degeneração das células do corno anterior na medula espinhal e núcleos motores na parte inferior do tronco cerebral. Existem 5 tipos clínicos de AME, cada um com uma apresentação clínica distinta, sendo a fraqueza motora a característica comum. As apresentações mais MAIS Androgen Insensitivity Syndrome precoces estão associadas a fraqueza motora mais MAIS Androgen Insensitivity Syndrome grave, afetando a capacidade da criança de atingir os marcos de desenvolvimento de sentar ou de andar. Nos tipos mais MAIS Androgen Insensitivity Syndrome graves, respirar e engolir também podem tornar-se difíceis à medida que a doença progride. O prognóstico da AME é reservado. Nos tipos menos graves, os adultos têm uma esperança de vida normal. O diagnóstico inicial é clínico e é confirmado por testes Testes Gonadal Hormones genéticos. O tratamento é sobretudo de suporte, embora estejam a ser desenvolvidas novas terapêuticas. O prognóstico depende do tipo clínico.
Last updated: Dec 15, 2025
A atrofia muscular espinhal (AME) é um espectro de síndromes autossómicas recessivas caracterizadas por fraqueza e atrofia muscular proximal progressiva, possivelmente devido à degeneração das células do corno anterior na medula espinhal e núcleos motores na parte inferior do tronco cerebral.
A atrofia muscular espinhal apresenta-se como uma síndrome de lesão do neurónio motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology inferior com fraqueza muscular, que é mais MAIS Androgen Insensitivity Syndrome grave nos tipos 0, 1 e 2.
Deve suspeitar-se de atrofia muscular espinhal em qualquer bebé com fraqueza inexplicada ou hipotonia. O diagnóstico definitivo requer teste genético.
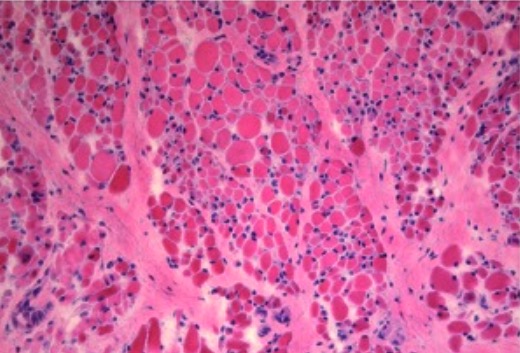
Biópsia muscular aos 10 meses de idade (H&E, × 200) com fibrose perimisial e endomisial acentuada, infiltração adipocitária em grupos de pequenas fibras e fibras hipertróficas dispersas
Imagem: “Muscle biopsy in SMA” por Beatriz San Millan et al. Licença: CC BY 2.5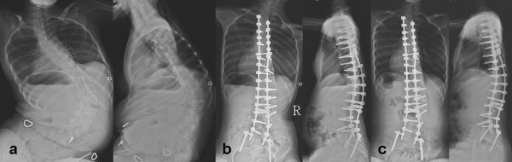
Imagens de uma menina de 13 anos com atrofia muscular espinhal (AME):
a: radiografia pré-operatória anteroposterior (AP) e lateral da coluna vertebral
b: radiografia pós-operatória AP e lateral da coluna vertebral
c: radiografia de seguimento AP e lateral da coluna
O tratamento atual é sobretudo de suporte e multidisciplinar (incluindo cuidados paliativos).
Terapêutica genética: já estão disponíveis 3 agentes nos EUA.
