Hyperkalemia is defined as a serum potassium (K+) concentration > 5.2 mEq/L. Homeostatic mechanisms maintain the serum K+ concentration between 3.5 and 5.2 mEq/L, despite marked variation in dietary intake. Hyperkalemia can be due to a variety of causes, which include transcellular Transcellular The movement of one cell into, through, and out of another cell. Tubular System shifts, tissue breakdown, inadequate renal excretion, and drugs. Hyperkalemia is usually asymptomatic if minor in severity; however, acute elevations or severe hyperkalemia can lead to potentially fatal cardiac arrhythmias. Management is guided by severity and includes measures to stabilize the myocardial membrane potential Membrane potential The membrane potential is the difference in electric charge between the interior and the exterior of a cell. All living cells maintain a potential difference across the membrane thanks to the insulating properties of their plasma membranes (PMs) and the selective transport of ions across this membrane by transporters. Membrane Potential, transiently shifting K+ intracellularly, removing K+ from the body, and treating the underlying predisposing conditions.
Last updated: Dec 15, 2025
K+ is the main intracellular cation in all cells and is distributed unevenly between the intracellular fluid Intracellular fluid The fluid inside cells. Body Fluid Compartments (98%) and extracellular fluid Extracellular fluid The fluid of the body that is outside of cells. It is the external environment for the cells. Body Fluid Compartments (2%).
A normal Western diet contains approximately 70–150 mmol of K+ per day. This diet is unlikely to lead to the development of hyperkalemia from increased intake only, owing to the following mechanisms:
The etiologies of hyperkalemia can be grouped into 5 categories: transcellular Transcellular The movement of one cell into, through, and out of another cell. Tubular System shifts, tissue breakdown, inadequate renal excretion, drug-induced, and pseudohyperkalemia.

Transcellular shift of K+:
Extracellular shift of K+:
1. Acidosis (increased H+) causes blockage of the Na+/H+ exchanger, which causes a decrease in intracellular Na+, in turn blocking Na+/K+ ATPase. On the other hand, acidosis activates the H+/K+ exchanger. Both cause an increase in extracellular K+.
2. Increased osmolarity in extracellular space (hyperglycemia, IV contrast, mannitol) shifts water outside the cell, decreasing K+ concentration. Increased gradient causes K+ diffusion outside.
Intracellular shift of K+:
1. Alkalosis (decreased H+) causes activation of the Na+/H+ exchanger, which causes an increase in intracellular Na+, in turn activating Na+/K+ ATPase. On the other hand, alkalosis blocks the H+/K+ exchanger. Both cause a decrease in extracellular K+.
2. Insulin and β2 adrenergic agonists activate Na+/K+ ATPase, lowering plasma K+ concentration.
Renal failure Renal failure Conditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate. Crush Syndrome:
Volume depletion Volume depletion Volume status is a balance between water and solutes, the majority of which is Na. Volume depletion refers to a loss of both water and Na, whereas dehydration refers only to a loss of water. Volume depletion can be caused by GI losses, renal losses, bleeding, poor oral Na intake, or third spacing of fluids. Volume Depletion and Dehydration:
Functional hypoaldosteronism Hypoaldosteronism Hypoaldosteronism is a hormonal disorder characterized by low levels of aldosterone. These low levels can be caused by decreased aldosterone production or a peripheral resistance to aldosterone. When hypoaldosteronism occurs as a result of an acquired decrease in renin production, the condition is more commonly referred to as renal tubular acidosis (RTA) type 4. Hypoaldosteronism:
Drugs are a very common cause of hyperkalemia and cause it by a variety of the previously mentioned mechanisms. A key part of the diagnosis of hyperkalemia is to review all the recent drugs and medications that a patient has received.
| Medication class (examples) | Mechanism |
|---|---|
| ACEi ACEi A class of drugs whose main indications are the treatment of hypertension and heart failure. They exert their hemodynamic effect mainly by inhibiting the renin-angiotensin system. They also modulate sympathetic nervous system activity and increase prostaglandin synthesis. They cause mainly vasodilation and mild natriuresis without affecting heart rate and contractility. Renin-Angiotensin-Aldosterone System Inhibitors (e.g., lisinopril Lisinopril One of the angiotensin-converting enzyme inhibitors (ACE inhibitors), orally active, that has been used in the treatment of hypertension and congestive heart failure. Renin-Angiotensin-Aldosterone System Inhibitors, captopril Captopril A potent and specific inhibitor of peptidyl-dipeptidase a. It blocks the conversion of angiotensin I to angiotensin II, a vasoconstrictor and important regulator of arterial blood pressure. Captopril acts to suppress the renin-angiotensin system and inhibits pressure responses to exogenous angiotensin. Hypertension Drugs) | Inhibits angiotensin II Angiotensin II An octapeptide that is a potent but labile vasoconstrictor. It is produced from angiotensin I after the removal of two amino acids at the c-terminal by angiotensin converting enzyme. The amino acid in position 5 varies in different species. To block vasoconstriction and hypertension effect of angiotensin II, patients are often treated with ace inhibitors or with angiotensin II type 1 receptor blockers. Renal Sodium and Water Regulation formation → decreases aldosterone secretion Secretion Coagulation Studies → decreases renal K+ secretion Secretion Coagulation Studies |
| ARB (e.g., losartan Losartan An antagonist of angiotensin type 1 receptor with antihypertensive activity due to the reduced pressor effect of angiotensin II. Hypertension Drugs, valsartan Valsartan A tetrazole derivative and angiotensin II type 1 receptor blocker that is used to treat hypertension. Hypertension Drugs) | Blocks angiotensin receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors → ↓ aldosterone secretion Secretion Coagulation Studies → ↓ renal K+ secretion Secretion Coagulation Studies |
| Direct renin inhibitors Direct Renin Inhibitors Renin-Angiotensin-Aldosterone System Inhibitors (e.g., aliskiren Aliskiren Renin-Angiotensin-Aldosterone System Inhibitors) | Blocks renin Renin A highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system. Renal Sodium and Water Regulation from converting angiotensinogen to angiotensin l → decreases aldosterone secretion Secretion Coagulation Studies → ↓ renal K+ excretion |
| K+-sparing diuretics Diuretics Agents that promote the excretion of urine through their effects on kidney function. Heart Failure and Chronic Coronary Syndrome Medication (e.g., amiloride Amiloride A pyrazine compound inhibiting sodium reabsorption through sodium channels in renal epithelial cells. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with diuretics to spare potassium loss. Liddle Syndrome, triamterene Triamterene A pteridinetriamine compound that inhibits sodium reabsorption through sodium channels in renal epithelial cells. Potassium-sparing Diuretics, spironolactone Spironolactone A potassium sparing diuretic that acts by antagonism of aldosterone in the distal renal tubules. It is used mainly in the treatment of refractory edema in patients with congestive heart failure, nephrotic syndrome, or hepatic cirrhosis. Its effects on the endocrine system are utilized in the treatments of hirsutism and acne but they can lead to adverse effects. Potassium-sparing Diuretics) | Block epithelial sodium channel Epithelial sodium channel Sodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure. Liddle Syndrome ( ENaC ENaC Sodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure. Liddle Syndrome) ( amiloride Amiloride A pyrazine compound inhibiting sodium reabsorption through sodium channels in renal epithelial cells. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with diuretics to spare potassium loss. Liddle Syndrome, triamterene Triamterene A pteridinetriamine compound that inhibits sodium reabsorption through sodium channels in renal epithelial cells. Potassium-sparing Diuretics) or the aldosterone receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors ( spironolactone Spironolactone A potassium sparing diuretic that acts by antagonism of aldosterone in the distal renal tubules. It is used mainly in the treatment of refractory edema in patients with congestive heart failure, nephrotic syndrome, or hepatic cirrhosis. Its effects on the endocrine system are utilized in the treatments of hirsutism and acne but they can lead to adverse effects. Potassium-sparing Diuretics, eplerenone Eplerenone A spironolactone derivative and selective aldosterone receptor antagonist that is used in the management of hypertension and congestive heart failure, post-myocardial infarction. Potassium-sparing Diuretics) → ↓ renal K+ excretion |
| Cardiac glycosides Cardiac glycosides Cardiac glycosides are a class of drugs reversibly inhibiting the sodium-potassium ATPase pump in myocardial cells and increasing vagal tone, which results in increased cardiac contractility and slowed conduction through the atrioventricular node. Cardiac Glycosides ( digoxin Digoxin A cardiotonic glycoside obtained mainly from digitalis lanata; it consists of three sugars and the aglycone digoxigenin. Digoxin has positive inotropic and negative chronotropic activity. It is used to control ventricular rate in atrial fibrillation and in the management of congestive heart failure with atrial fibrillation. Its use in congestive heart failure and sinus rhythm is less certain. The margin between toxic and therapeutic doses is small. Cardiac Glycosides) | Inhibits Na+/K+ ATPase pump Pump ACES and RUSH: Resuscitation Ultrasound Protocols → less K+ moved into cells |
| NSAIDs NSAIDS Primary vs Secondary Headaches (e.g., ibuprofen Ibuprofen A nonsteroidal anti-inflammatory agent with analgesic properties used in the treatment of rheumatism and arthritis. Nonsteroidal Antiinflammatory Drugs (NSAIDs)) | Decreases renin Renin A highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system. Renal Sodium and Water Regulation and aldosterone → ↓ renal K+ secretion Secretion Coagulation Studies |
| Calcineurin inhibitors Calcineurin Inhibitors Compounds that inhibit or block the phosphatase activity of calcineurin. Immunosuppressants (e.g., cyclosporine Cyclosporine A cyclic undecapeptide from an extract of soil fungi. It is a powerful immunosupressant with a specific action on T-lymphocytes. It is used for the prophylaxis of graft rejection in organ and tissue transplantation. Immunosuppressants, tacrolimus Tacrolimus A macrolide isolated from the culture broth of a strain of streptomyces tsukubaensis that has strong immunosuppressive activity in vivo and prevents the activation of T-lymphocytes in response to antigenic or mitogenic stimulation in vitro. Immunosuppressants) | Multifactorial/incompletely understood: ↓ aldosterone release, ↓ aldosterone sensitivity, inhibition of Na+/K+ ATPase pump Pump ACES and RUSH: Resuscitation Ultrasound Protocols, blocking of ENaC ENaC Sodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure. Liddle Syndrome channel |
| Succinylcholine Succinylcholine A quaternary skeletal muscle relaxant usually used in the form of its bromide, chloride, or iodide. It is a depolarizing relaxant, acting in about 30 seconds and with a duration of effect averaging three to five minutes. Succinylcholine is used in surgical, anesthetic, and other procedures in which a brief period of muscle relaxation is called for. Cholinomimetic Drugs | Causes extracellular leakage of K+ through acetylcholine Acetylcholine A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system. Receptors and Neurotransmitters of the CNS receptor-gated channels Channels The Cell: Cell Membrane |
| Antimicrobials (e.g., trimethoprim Trimethoprim The sulfonamides are a class of antimicrobial drugs inhibiting folic acid synthesize in pathogens. The prototypical drug in the class is sulfamethoxazole. Although not technically sulfonamides, trimethoprim, dapsone, and pyrimethamine are also important antimicrobial agents inhibiting folic acid synthesis. The agents are often combined with sulfonamides, resulting in a synergistic effect. Sulfonamides and Trimethoprim, pentamidine) | Block ENaC ENaC Sodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure. Liddle Syndrome channel |
The most severe symptoms of hyperkalemia are impaired electrical conduction in the heart. Cardiac symptoms are more likely to occur with increasing severity and acuity of hyperkalemia; however, even relatively severe hyperkalemia can be asymptomatic. Muscular symptoms may be observed, and these include weakness and paralysis.
Cardiac symptoms are the most important symptoms of hyperkalemia, as they can be rapidly fatal.
Some patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship will not have ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) changes or arrhythmias, even with severe hyperkalemia.
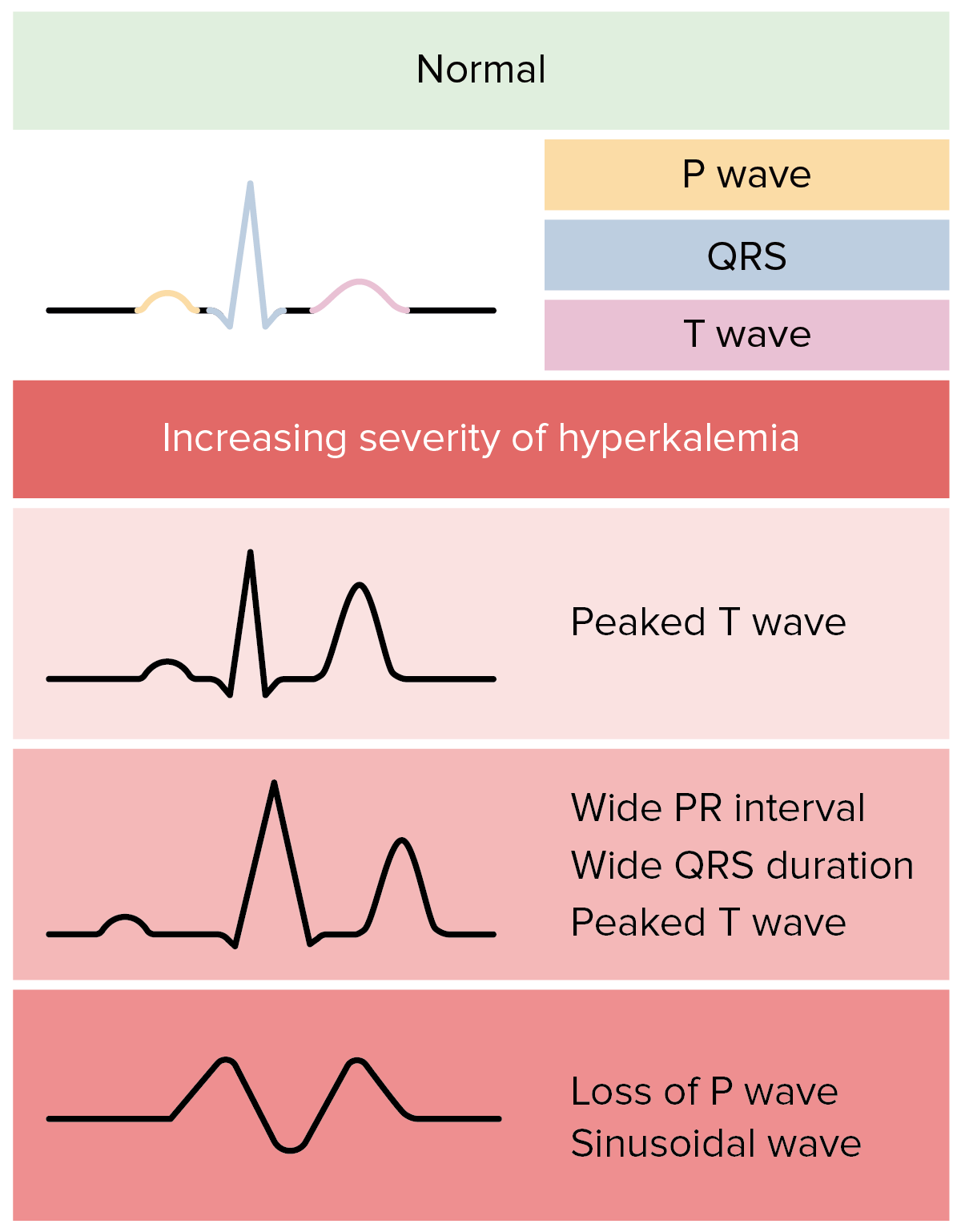
ECG changes in hyperkalemia:
In reality, ECG changes in hyperkalemia are more variable and less predictable.
The management of hyperkalemia often takes precedence over the diagnosis because of the possibility of life-threatening arrhythmias and is guided by determining the level of urgency needed for treatment. Usually, the etiology of hyperkalemia is not difficult to determine and is not impeded by treating it first.
| Goal | Intervention | Properties/indication |
|---|---|---|
| Stabilize the myocardium Myocardium The muscle tissue of the heart. It is composed of striated, involuntary muscle cells connected to form the contractile pump to generate blood flow. Heart: Anatomy | IV calcium Calcium A basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes. Electrolytes |
|
| Shift K+ into cells | Insulin Insulin Insulin is a peptide hormone that is produced by the beta cells of the pancreas. Insulin plays a role in metabolic functions such as glucose uptake, glycolysis, glycogenesis, lipogenesis, and protein synthesis. Exogenous insulin may be needed for individuals with diabetes mellitus, in whom there is a deficiency in endogenous insulin or increased insulin resistance. Insulin |
|
| Sodium Sodium A member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23. Hyponatremia bicarbonate Bicarbonate Inorganic salts that contain the -HCO3 radical. They are an important factor in determining the ph of the blood and the concentration of bicarbonate ions is regulated by the kidney. Levels in the blood are an index of the alkali reserve or buffering capacity. Electrolytes (NaHCO3) |
|
|
| β2 agonist |
|
|
| Remove K+ from the body | Via urine |
|
| Via GI tract | Cation exchange resins: bind BIND Hyperbilirubinemia of the Newborn K+ in exchange for Na+ or Ca CA Condylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding. Condylomata Acuminata (Genital Warts)2+ | |
| Dialysis Dialysis Renal replacement therapy refers to dialysis and/or kidney transplantation. Dialysis is a procedure by which toxins and excess water are removed from the circulation. Hemodialysis and peritoneal dialysis (PD) are the two types of dialysis, and their primary difference is the location of the filtration process (external to the body in hemodialysis versus inside the body for PD). Peritoneal Dialysis and Hemodialysis |
|