Liddle syndrome, a type of pseudohyperaldosteronism, is a rare cause of secondary hypertension. Liddle syndrome results from autosomal dominant gain-of-function mutations in the genes that encode the epithelial sodium channel (ENaC) subunits, also known as the "collecting tubule sodium channel" or "amiloride-sensitive sodium channel." The activity of ENAC is increased, leading to sodium Sodium A member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23. Hyponatremia and water retention. Liddle syndrome presents with the classic triad of resistant hypertension Resistant hypertension Blood pressure that remains uncontrolled despite concurrent use of 3 antihypertensive agents of different classes. Uncontrolled Hypertension at an early age, hypokalemia Hypokalemia Hypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake. Hypokalemia, and metabolic alkalosis Alkalosis A pathological condition that removes acid or adds base to the body fluids. Respiratory Alkalosis, which mimics the symptoms of primary aldosteronism; however, this syndrome is associated with low plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products aldosterone Aldosterone A hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium. Hyperkalemia levels. Diagnosis is based on history and physical examination, blood and urine analysis, and genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Management is by using potassium-sparing diuretics Diuretics Agents that promote the excretion of urine through their effects on kidney function. Heart Failure and Chronic Coronary Syndrome Medication and restricting dietary sodium Sodium A member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23. Hyponatremia. Treatment delay can lead to serious cardiovascular and renal complications. The prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas is good if Liddle syndrome is treated early.
Last updated: Dec 15, 2025
Liddle syndrome is a rare autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance genetic disorder associated with abnormal increased function of epithelial sodium Sodium A member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23. Hyponatremia channels Channels The Cell: Cell Membrane (ENaC) in the collecting tubules Collecting tubules Straight tubes commencing in the radiate part of the kidney cortex where they receive the curved ends of the distal convoluted tubules. In the medulla the collecting tubules of each pyramid converge to join a central tube (duct of Bellini) which opens on the summit of the papilla. Kidneys: Anatomy. Liddle syndrome is clinically characterized by hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, low plasma renin activity Plasma renin activity Renal Artery Stenosis, metabolic alkalosis Alkalosis A pathological condition that removes acid or adds base to the body fluids. Respiratory Alkalosis, hypokalemia Hypokalemia Hypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake. Hypokalemia, and low aldosterone Aldosterone A hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium. Hyperkalemia levels.
An autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance gain-of-function gene mutation Gene Mutation Myotonic Dystrophies changes the structure of an ENaC subunit and also affects the region of the protein involved in signaling for its breakdown, which is normally controlled by aldosterone Aldosterone A hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium. Hyperkalemia.
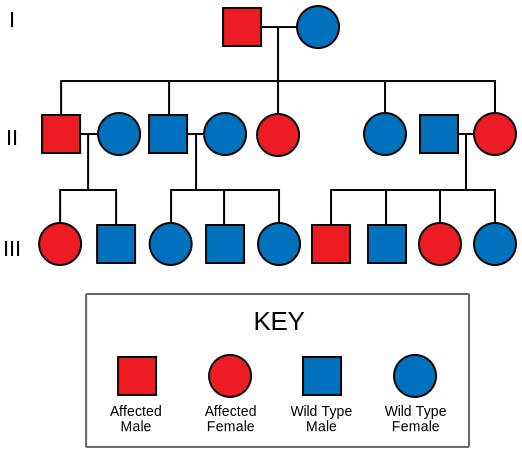
The autosomal dominant inheritance of Liddle syndrome
Image: “Liddle” by Dra marina. License: CC0 1.0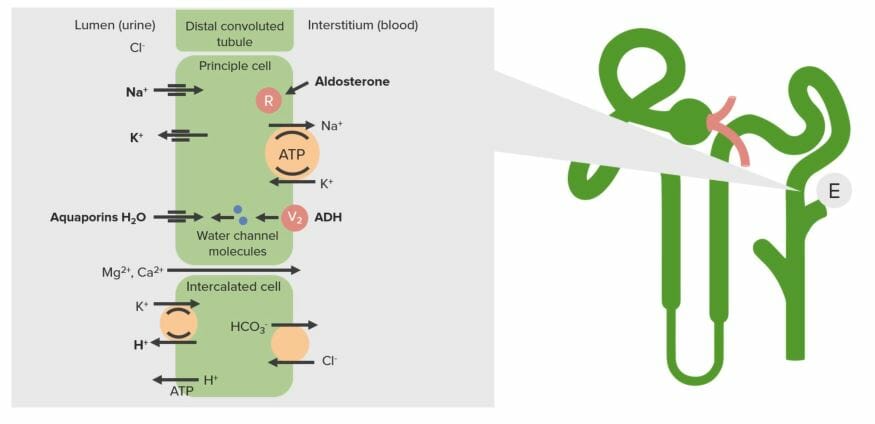
The transport system of the renal collecting duct (E):
In Liddle syndrome, an increase in sodium channels on the apical surface (lumen side) results in increased sodium reabsorption. This sodium is transported back to the blood via the Na/K ATPase on the basolateral side, resulting in an increase in intracellular potassium. This potassium is then secreted via potassium channels on the apical surface.
Liddle syndrome is rare and has a presentation similar to many other syndromes associated with mineralocorticoid excess Mineralocorticoid excess A hereditary disease characterized by childhood onset hypertension, hypokalemic alkalosis, and low renin and aldosterone secretion. It results from a defect in the activity of the 11-beta-hydroxysteroid dehydrogenase type 2 enzyme which results in inadequate conversion of cortisol to cortisone. The build up of unprocessed cortisol to levels that stimulate mineralocorticoid receptors creates the appearance of having excessive mineralocorticoids. Metabolic Alkalosis. Thus, Liddle syndrome is often misdiagnosed or remains undetected.
The classic triad of the presentation includes:
The preliminary diagnosis of Liddle syndrome is based mainly on suspicion resulting from the clinical presentation. It is also important to:
Laboratory findings:
Genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies should be considered in individuals with a family history Family History Adult Health Maintenance of Liddle syndrome and in those in whom this condition is suspected.
Early diagnosis of Liddle syndrome leads to resolution or control of the disorder, enabling the affected individual to lead a normal life.
Owing to its rare occurrence and similarity of symptoms to those of other disorders associated with mineralocorticoid excess Mineralocorticoid excess A hereditary disease characterized by childhood onset hypertension, hypokalemic alkalosis, and low renin and aldosterone secretion. It results from a defect in the activity of the 11-beta-hydroxysteroid dehydrogenase type 2 enzyme which results in inadequate conversion of cortisol to cortisone. The build up of unprocessed cortisol to levels that stimulate mineralocorticoid receptors creates the appearance of having excessive mineralocorticoids. Metabolic Alkalosis, Liddle syndrome is often misdiagnosed, which may cause an increase in the occurrence of complications: