DiGeorge syndrome (DGS) is a condition caused by a microdeletion at location q11.2 of chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics 22 (thus also called 22q11.2 deletion syndrome). There is a defective development of the third and fourth pharyngeal pouches Pharyngeal pouches Branchial Apparatus and Aortic Arches, leading to thymic and parathyroid Parathyroid The parathyroid glands are 2 pairs of small endocrine glands found in close proximity to the thyroid gland. The superior parathyroid glands are lodged within the parenchyma of the upper poles of the right and left thyroid lobes; the inferior parathyroid glands are close to the inferior tips or poles of the lobes. Parathyroid Glands: Anatomy hypoplasia Hypoplasia Hypoplastic Left Heart Syndrome (HLHS) (causing T-cell immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome and hypocalcemia Hypocalcemia Hypocalcemia, a serum calcium < 8.5 mg/dL, can result from various conditions. The causes may include hypoparathyroidism, drugs, disorders leading to vitamin D deficiency, and more. Calcium levels are regulated and affected by different elements such as dietary intake, parathyroid hormone (PTH), vitamin D, pH, and albumin. Presentation can range from an asymptomatic (mild deficiency) to a life-threatening condition (acute, significant deficiency). Hypocalcemia, respectively). Conotruncal anomalies presenting as congenital heart defects are also characteristic of the disease. Other manifestations consist of characteristic facial features, frequent infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, and neuropsychiatric disorders. Diagnosis is obtained by a combination of clinical findings, laboratory tests (reduced T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified - cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions and low calcium Calcium A basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes. Electrolytes), echocardiogram Echocardiogram Transposition of the Great Arteries, and genetic analysis. Treatment can include calcium Calcium A basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes. Electrolytes supplementation, prophylactic antibiotics, surgery (for heart defects and palate Palate The palate is the structure that forms the roof of the mouth and floor of the nasal cavity. This structure is divided into soft and hard palates. Palate: Anatomy abnormalities), and thymus Thymus A single, unpaired primary lymphoid organ situated in the mediastinum, extending superiorly into the neck to the lower edge of the thyroid gland and inferiorly to the fourth costal cartilage. It is necessary for normal development of immunologic function early in life. By puberty, it begins to involute and much of the tissue is replaced by fat. Lymphatic Drainage System: Anatomy or hematopoietic cell transplantation. Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas depends on the severity of cardiac anomaly and immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome.
Last updated: Dec 15, 2025
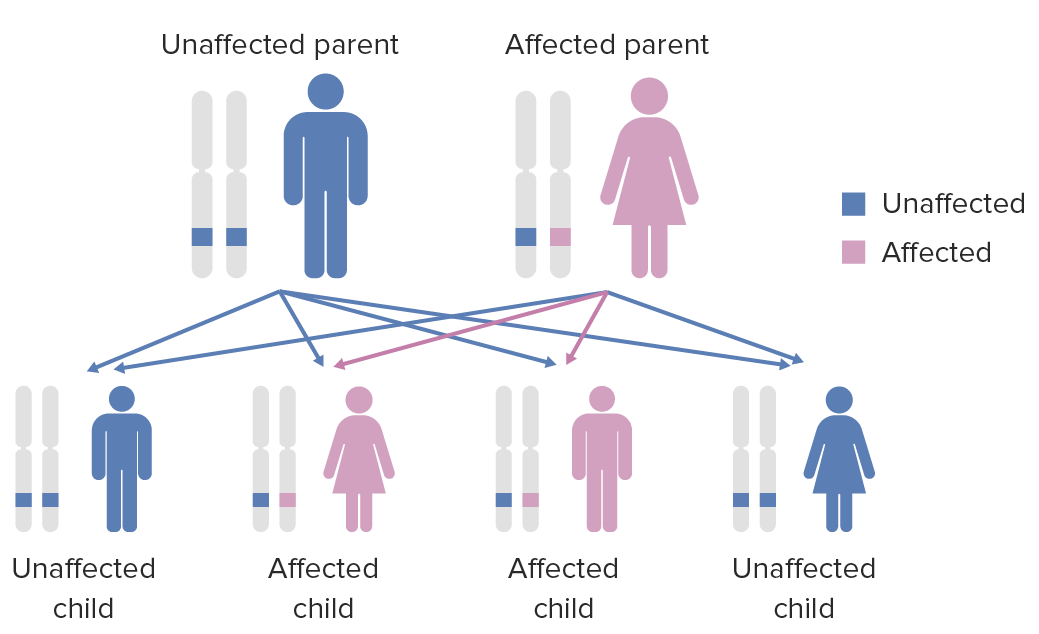
Diagram of the inheritance pattern of autosomal dominant conditions
Image by Lecturio.
DiGeorge syndrome: deletion at 11.2 of q arm
Image by Lecturio.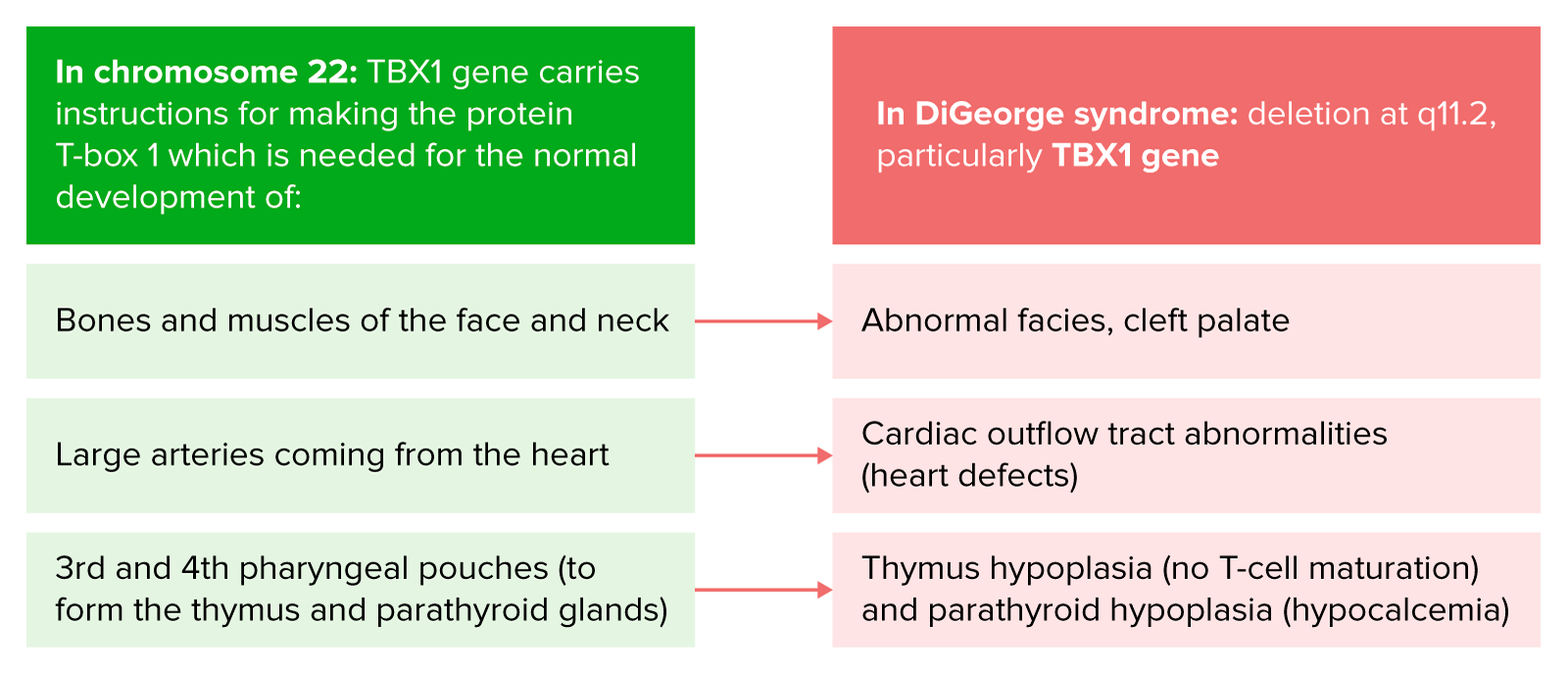
Pathophysiology of DiGeorge syndrome
Image by Lecturio.DiGeorge syndrome has a marked variability in clinical expression among different individuals. Manifestations can include the following:
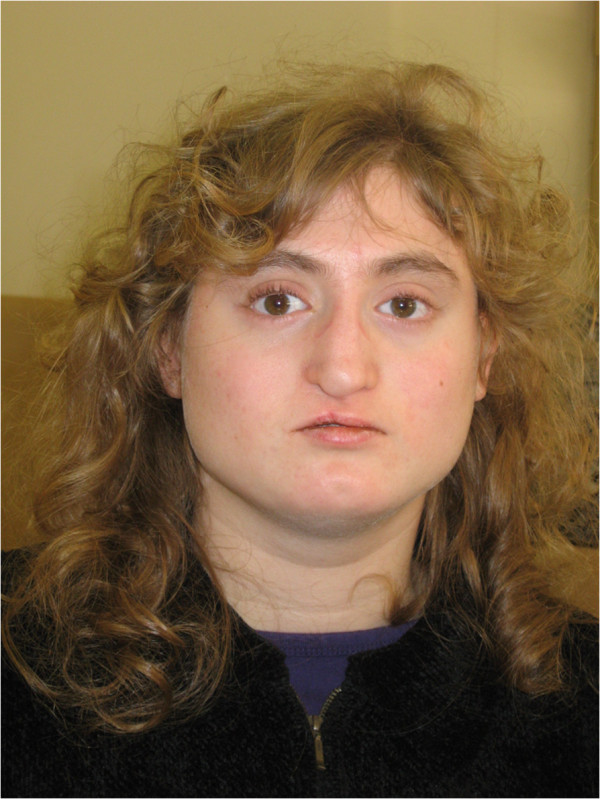
Facial appearance of a patient with atypical 22q11.2 deletion. Note the broad, square face; hypotelorism; narrow nasal base and broad nasal bridge; overhanging nasal tip; deviated nasal tip (state post cleft lip repair); short philtrum; narrow mouth; chin dimple; and broad neck.
Image: “Facial appearance of Patient VC901” by Elena Michaelovsky et al. License: CC BY 2.0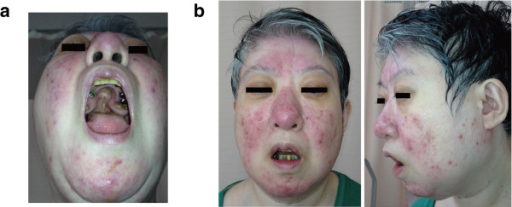
22q11.2 deletion features in a 48-year-old woman.
(a) Cleft palate; had previous surgery
(b) Mild dysmorphic facial features, including a low anterior hairline, swollen eyelids, malar flatness, nose with a bulbous nasal tip, hypoplastic nasal alae and a square and flat nasal root, small mouth, and a thin upper lip
DiGeorge syndrome signs can be summarized using the mnemonic CATCH-22:
Establishing a diagnosis is difficult due to the variability of phenotypes. A diagnosis of DiGeorge is determined by demonstrating a decrease in CD3 CD3 Complex of at least five membrane-bound polypeptides in mature T-lymphocytes that are non-covalently associated with one another and with the T-cell receptor. The CD3 complex includes the gamma, delta, epsilon, zeta, and eta chains (subunits). When antigen binds to the T-cell receptor, the CD3 complex transduces the activating signals to the cytoplasm of the T-cell. The CD3 gamma and delta chains (subunits) are separate from and not related to the gamma/delta chains of the T-cell receptor. T cells: Types and Functions+ T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified – cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions in addition to characteristic clinical findings and genetic studies demonstrating deletion in chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics area 22q11.2.
Management is aimed at treating the associated features of the disease. Early intervention and developmental evaluation are key.
There is no cure for DiGeorge syndrome. Life expectancy Life expectancy Based on known statistical data, the number of years which any person of a given age may reasonably expected to live. Population Pyramids largely depends on the degree of cardiac defects (most important factor) and immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome.