El síndrome de DiGeorge es una afección causada por una microdeleción en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la ubicación q11.2 del cromosoma 22 (también llamado síndrome de deleción 22q11.2). Hay un desarrollo defectuoso de la tercera y cuarta bolsas faríngeas, lo que conduce a hipoplasia tímica y paratiroidea (que causa inmunodeficiencia de células T e hipocalcemia, respectivamente). Las anomalías conotruncales que se presentan como defectos cardíacos congénitos también son características de la enfermedad. Otras manifestaciones consisten en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum rasgos faciales característicos, infecciones frecuentes y trastornos neuropsiquiátricos. El diagnóstico se obtiene mediante una combinación de hallazgos clínicos, pruebas de laboratorio (células T reducidas y calcio bajo), ecocardiograma y análisis genético. El tratamiento puede incluir suplementos de calcio, antibióticos profilácticos, cirugía (para defectos cardíacos y anomalías del paladar) y trasplante de timo o células hematopoyéticas. El pronóstico depende de la gravedad de la anomalía cardíaca y la inmunodeficiencia.
Last updated: Dec 15, 2025
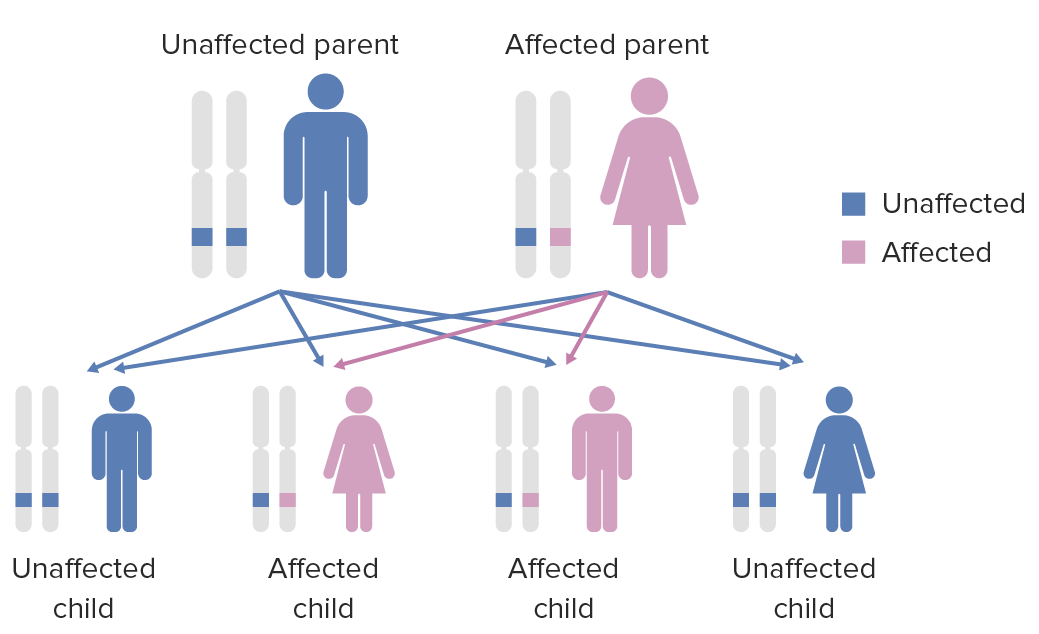
Diagrama del patrón de herencia de las condiciones autosómicas dominantes
Imagen por Lecturio.
Síndrome de DiGeorge: deleción en 11.2 del brazo q
Imagen por Lecturio.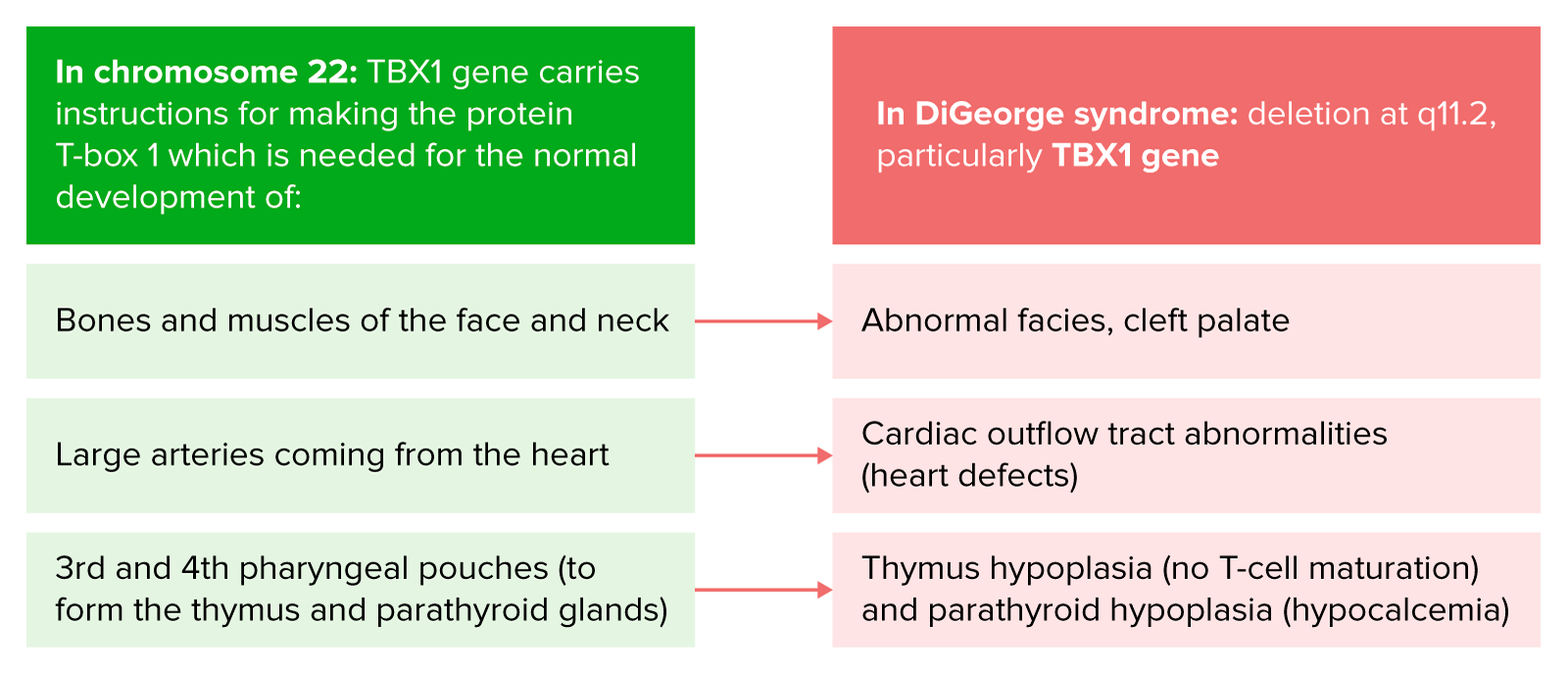
Fisiopatología del síndrome de DiGeorge
Imagen por Lecturio.El síndrome de DiGeorge tiene una marcada variabilidad en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la expresión clínica entre diferentes individuos. Las manifestaciones pueden incluir las siguientes:
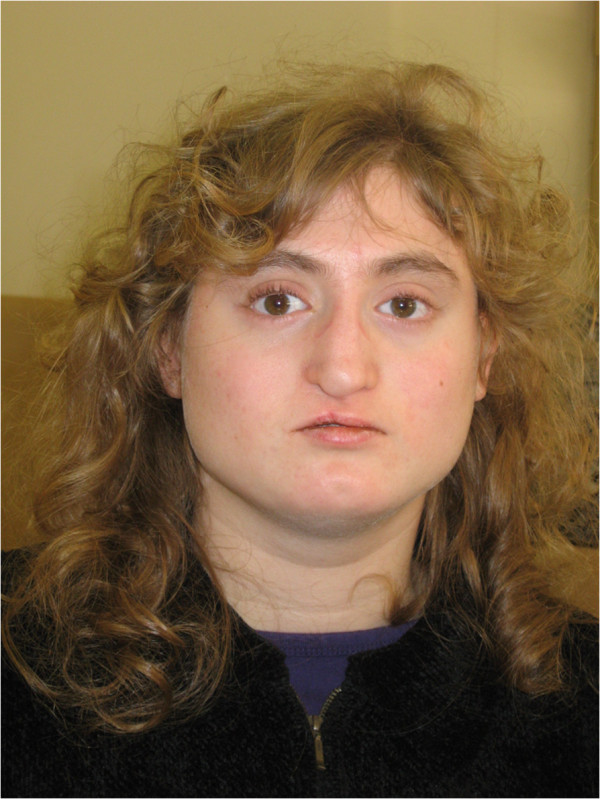
Aspecto facial de una paciente con deleción 22q11.2 atípica. Es de notar la cara ancha y cuadrada; hipotelorismo; base nasal estrecha y puente nasal ancho; punta nasal sobresaliente; punta nasal desviada (estado posterior a la reparación del labio leporino); surco nasolabial corto; boca estrecha hoyuelo en la barbilla; y cuello ancho.
Imagen: “Facial appearance of Patient VC901” por Elena Michaelovsky et al. Licencia: CC BY 2.0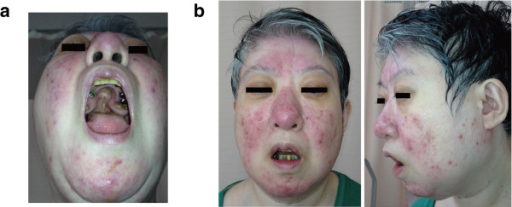
Características de la deleción 22q11.2 en una mujer de 48 años.
(a) Paladar hendido; tuvo una cirugía previa
(b) Rasgos faciales dismórficos leves, que incluyen una línea del cabello anterior baja, párpados hinchados, planitud malar, nariz con una punta nasal bulbosa, alas nasales hipoplásicas y una raíz nasal cuadrada y plana, boca pequeña y un labio superior delgado
Los LOS Neisseria signos del síndrome de DiGeorge se pueden resumir utilizando el mnemónico CATCH-22 ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés):
Establecer un diagnóstico es difícil debido a la variabilidad de los LOS Neisseria fenotipos. El diagnóstico de DiGeorge se determina al AL Amyloidosis demostrar una disminución de las células T CD3 CD3 Complex of at least five membrane-bound polypeptides in mature T-lymphocytes that are non-covalently associated with one another and with the T-cell receptor. The CD3 complex includes the gamma, delta, epsilon, zeta, and eta chains (subunits). When antigen binds to the T-cell receptor, the CD3 complex transduces the activating signals to the cytoplasm of the T-cell. The CD3 gamma and delta chains (subunits) are separate from and not related to the gamma/delta chains of the T-cell receptor. T cells: Types and Functions+, además de los LOS Neisseria hallazgos clínicos característicos y los LOS Neisseria estudios genéticos que demuestran la deleción en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el área cromosómica 22q11.2.
El tratamiento tiene como objetivo tratar las características asociadas de la enfermedad. La intervención temprana y la evaluación del desarrollo son fundamentales.
No existe cura para el síndrome de DiGeorge. La esperanza de vida depende en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum gran medida del grado de defectos cardíacos (factor más importante) y la inmunodeficiencia.